


许多疾病中都报告了肠道微生物组的变化(Duvallet等人,2017年;Pasolli等人,2016年)。识别这些变化的特异性对于开发基于微生物组的诊断和治疗策略至关重要。除了调查因果关系之外,微生物组-疾病变化的三个关键方面需要阐明(Duvallet等人,2017年;He等人,2018年)。第一个方面涉及与疾病相关的组成部分(关键分类群及其代谢功能),这将有助于建立诊断并理解改变的微生物组如何影响宿主。第二个方面涉及关联的方向性,这对于设计治疗策略是必需的(例如,针对性的抗微生物治疗与微生物组恢复)。第三个方面是共享与疾病特异性微生物组变化的程度,这表明某种特定疾病的发病可能对其他疾病的微生物组相关风险产生什么影响。跨不同地理位置的病例和对照的元分析已经确定了多种疾病共有的微生物组变化成分,以及疾病特异性变化(Duvallet等人,2017年;He等人,2018年;Jackson等人,2018年;Pasolli等人,2016年)。虽然某些疾病如结直肠癌(CRC)的特点是病原菌(如梭杆菌属、拟杆菌属、小杆菌属)的增加(或获得),其他疾病如炎症性肠病(IBD)的发病与特定分类群的耗竭有关(例如,玫瑰杆菌属、粪便杆菌属)(Duvallet等人,2017年)。相比之下,腹泻疾病伴随着病原菌(特别是肠杆菌科)的增加以及共生分类群的丰度降低。最近的元分析还显示出微生物组关联的惊人重叠程度(无论是在特定分类群的水平还是特定微生物代谢途径的水平)(Armour等人,2019年;Duvallet等人,2017年;Pasolli等人,2016年)。尽管存在这些重叠,但微生物分类群与在多种疾病中始终发生变化的功能途径之间的联系尚未详细探讨。

人体是一个由人类细胞和数万亿微生物组成的生态系统,最大的微生物群落位于肠道。这个肠道微生物群落帮助我们从食物中收获营养,调节我们的免疫系统,甚至影响我们的情绪。传染性和慢性疾病似乎会导致肠道微生物组的组成发生变化,而微生物组的变化可能增加一些非传染性疾病的风险。因此,更多地了解与疾病相关的肠道微生物组的变化可能有助于科学家开发新的检测和治疗方法。为此,科学家需要了解哪些微生物在个别疾病中起作用,与风险相关的微生物是在患者身上获得还是有益微生物的丧失,以及是否有许多疾病中肠道微生物的特定变化发生。
衰老也会改变肠道微生物。这可能是因为老年人饮食不太复杂,并且可能服用许多可能改变他们肠道微生物的药物。因此,年龄可能会影响与疾病相关的肠道微生物的变化。这突显了需要研究来区分衰老相关和疾病相关变化在肠道微生物组中的重要性。
现在,Ghosh等人表明,与疾病相关的肠道微生物的变化可能随个人的年龄而变化。该分析比较了20至89岁超过2500名个体的肠道微生物组。这包括了患有炎症性肠病、结直肠癌、2型糖尿病、肠道息肉和肝硬化的个体。研究揭示了年轻人逐渐获得与疾病相关的肠道微生物,而老年人倾向于失去通常在健康肠道中找到的肠道微生物。Ghosh等人还识别了一组在许多疾病和年龄组中获得的肠道微生物。这组微生物也与老年人的虚弱有关。这组微生物的特征都是已知对人类健康有害的。
这项分析显示了控制年龄和其他可能歪曲微生物组项目结果的因素的重要性。未来的研究需要了解这些肠道微生物变化为何发生,以及这些变化对个人健康和疾病进程的后果是什么。这可能导致开发出帮助促进健康肠道微生物组和抵抗一生疾病的治疗方法。基于微生物组的诊断/治疗还需要考虑几个宿主因素。受试者的地区/种族变异与肠道微生物组组成和多种疾病中的基于微生物组的诊断有关(Deschasaux等人,2018年;He等人,2018年)。除了地区/种族特异性变异外,多项研究还表明年龄是微生物组组成的强协变量(Falony等人,2016年)。我们和其他研究者先前已经在ELDERMET队列中识别了与衰老相关的微生物组变化,这些变化与较低的复杂性饮食摄入和多种药物治疗有关(Claesson等人,2012年;O’Toole和Jeffery,2015年;Ticinesi等人,2017年)。
除了衰老相关的微生物组变化外,不同疾病的早期和晚期变异版本具有不同的病理生理学特征(Duricova等人,2014年;Yeo等人,2017年),这也可能与衰老相关的微生物组参与有关。这些观察激发了我们在人类年龄景观中对微生物组-疾病变化进行广泛的调查。
我们的目标是回答以下问题:衰老在多大程度上影响不同疾病的微生物组-疾病关联?不同疾病在不同年龄组中是否显示出不同的微生物组特征(无论是在微生物组组成部分方面还是在变化方向性方面)?如果是这样,这些变化在多大程度上影响了已知的基于微生物组的标记物?在不同年龄组中,是否出现了多种与疾病相关的分类群的模式变化(如先前的元分析所识别的)?最后,是否有可能识别出这些疾病标记物与先前与这些疾病相关的微生物代谢途径之间的联系? 在广泛的年龄范围内调查多种疾病中的微生物组变化需要一个包含一致整理的微生物组剖面的全面数据集。ExperimentHub R库的curated MetagenomicData存储库包含了来自不同人体部位的6000多个宏基因组测序的人类微生物组样本的分类学和功能途径剖面,涵盖来自25多项不同研究的30多种疾病(和对照)(Pasolli等人,2017年)。curated MetagenomicData的另一个优点是这些微生物组剖面是使用统一的生物信息学分析创建的(使用metaphlan2和humann2)(Franzosa等人,2018年;Truong等人,2015年)。该存储库还为样本提供了广泛的元数据信息,包括研究、实验协议、地区(或国家)、疾病状态、年龄、性别、BMI和抗生素使用情况。尽管其他影响肠道微生物组组成的关键因素,如饮食或药物,在所有研究中并未提供,但该存储库仍能促进对多项研究的数据集进行元分析,以提供初步见解,了解其他关键宿主相关因素对微生物组疾病特征的影响。在这里,我们分析了来自15项研究的肠道微生物组,包括四种疾病,涵盖从20岁到89岁的2500多名个体,这些数据来自ExperimentHub存储库、ELDERMET项目以及最近发表的关于IBD和结直肠癌CRC的研究(Franzosa等人,2019年;Thomas等人,2019年;Wirbel等人,2019年)。

我们首先采用逐步方法来减少不同研究中DNA测序/提取方法对微生物组剖面造成的混淆效应(见材料和方法;图1—图补充1),随后保留了20至89岁年龄范围内的个体样本(排除了“对照组”也由住院患者组成的队列)(Vincent等人,2016年)。我们通过补充来自最近发表的关于IBD(Franzosa等人,2019年)和CRC(作为“验证”队列)(Thomas等人,2019年;Wirbel等人,2019年)的475个射枪宏基因组剖面的数据集,最终组装了一个超过2500个样本的微生物组分类剖面的综合集(包括后来在研究中使用的189个ELDERMET样本)(见材料和方法;补充文件1和补充文件2;图1—图补充2)。
接下来,我们研究了元数据与分类学剖面的相互作用。在过滤掉冗余和稀疏的元数据类型(记录的样本少于30%)后,我们进行了PERMANOVA分析,将每种元数据的效果与肠道微生物组联系起来,同时考虑DNA提取技术作为一个混杂因素(见材料和方法;DNA提取技术仍被视为混杂因素,因为它对肠道微生物组的组成仍有轻微影响,R2 = 0.019)。地区因素,即国家和大洲,与肠道微生物组组成有最大的交互作用(图1A)。地区因素反映了研究人群的种族和其他社会经济属性,这对肠道微生物组的结构和基于微生物组的疾病特征有主导影响(Deschasaux等人,2018年;He等人,2018年)。其次是年龄,然后是研究状况(疾病与对照状态)。因此,我们使用主成分分析法(PCA)探索了年龄范围内明显“健康”的微生物组的变化,分析了来自20-39岁(归类为“年轻”)、40-59岁(“中年”)和60岁以上(“老年”)年龄组的1175个肠道微生物组剖面,这些样本均来自“对照”个体。在调整国家和DNA提取技术后进行的肠道微生物组组成的PERMANOVA分析表明,老年对照组与年轻和中年个体相比,具有显著不同的微生物组组成(p<0.001)(图1B),这与我们之前的发现一致(Claesson等人,2012年)。为了进一步探索这种效应,我们随后研究了五个疾病(IBD、II型糖尿病[T2D]、肠息肉、CRC和肝硬化)中受试者年龄对肠道微生物组组成的影响,这些疾病在至少两个年龄组中有大量病例可用。不同疾病对应的样本可在不同的队列中获得(表1)。每个队列数据集包括健康对照组和疾病个体,根据地理区域和环境匹配(从而减少这些对肠道微生物组的混杂效应),因此我们可以进行PERMANOVA分析,量化年龄组(“年轻”、“中年”和“老年”)和疾病状态(“对照”和“疾病”)对肠道微生物组的影响(在每个不同的疾病特异性队列中分别进行)。对于所有五种疾病,年龄都有显著影响。值得注意的是,对于三种疾病(T2D、CRC和肠息肉),这种效应高于疾病本身的影响(图1C)。
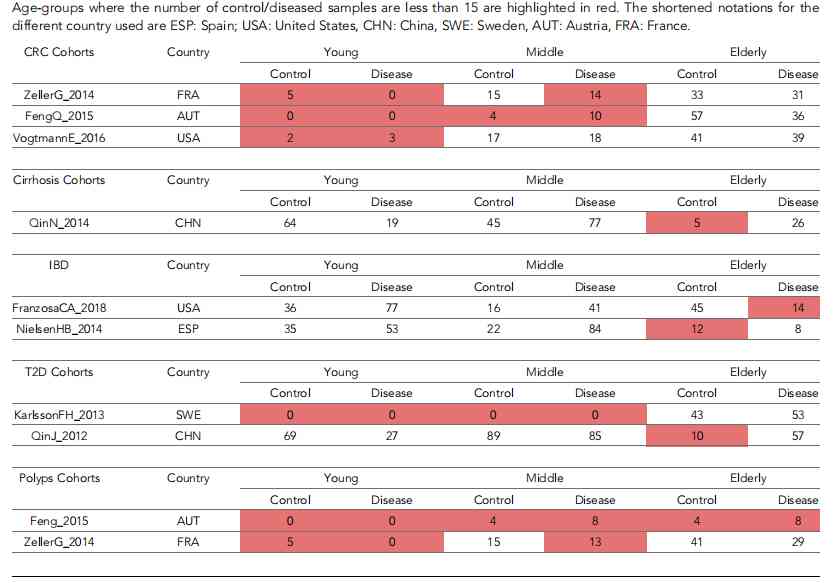
表1. 属于与每种疾病相关的大陆特定组中不同年龄组的对照和患病个体数量
接下来,我们调查了这些与年龄相关的微生物组组成变化是否影响了疾病特征。然而,进行这项调查带来了一个特定的挑战。鉴于像国家或大洲这样的地区因素对肠道微生物组组成有最高的影响,任何对不同疾病的年龄组特异性微生物组特征的调查都需要在地理上同质的亚人群(或研究队列)内进行比较,以确保任何疾病特征的差异不仅仅是由地区肠道微生物组组成的变异所驱动的。然而,专注于特定疾病队列(属于特定国家)也带来了限制,包括样本量减少(因此统计效力降低)和对某些年龄组的偏见。在我们这里分析的八个疾病队列(在curateMetagenomicData中)中,我们发现其中一半显示出对照个体和患病个体的年龄存在显著差异(即它们没有年龄匹配)(图1D)。此外,大多数疾病队列中特定年龄组的代表性也存在偏差。通过增加来自与相应疾病队列相同的地区(国家或大洲)的其他队列中的“对照”样本,预期可以通过增加可用样本的数量,从而增加比较的统计效力,来规避这个问题。我们通过将样本分入疾病特异性的国别/大洲级别的箱子中来解决这个问题(见材料和方法;图1—图补充3)。 这确保了所有疾病中三个不同年龄组的样本有足够的代表性。
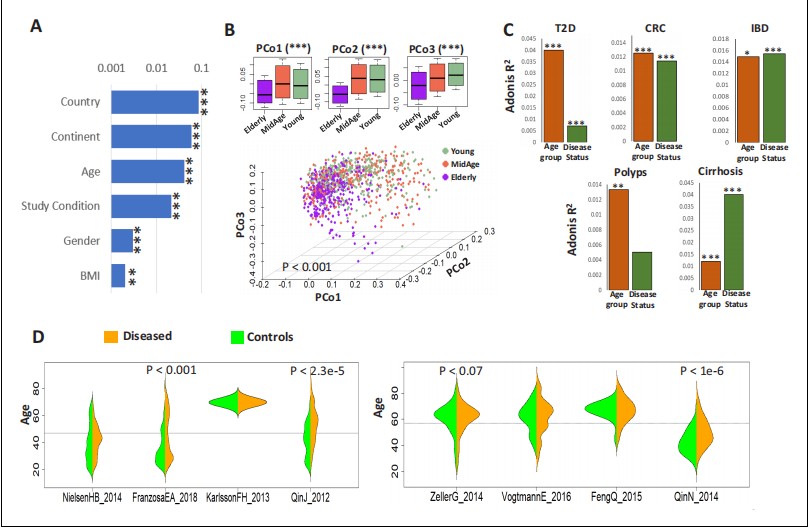
图1. 年龄影响微生物组组成以及微生物组-疾病特征。 (A) 条形图显示了宿主因素对ExperimentHub存储库中微生物组组成的影响(通过使用PERMANOVA计算R2值,调整DNA提取技术作为混杂因素后得出)。仅显示至少30%样本可用的元数据。关联显著性的p值也标示出来,****: p<0.0001; ***: p<0.001, **: p<0.01, *: p<0.05。(B) 主坐标分析(PCoA)图展示了按三个年龄范围分组的“对照”样本的物种剖面,青年(20-39岁)、中年(40-59岁)和老年(60岁及以上)。使用PERMANOVA(adonis)计算了三组之间的差异的显著性(p值),同时考虑了特定国家的差异和DNA提取技术,也标示出来。顶部的箱形图显示了属于三个年龄组的样本的前三个PCoA坐标的变化。老年个体的微生物组与年轻/中年个体显著不同。(C) PERMANOVA R2值的条形图显示了五个疾病队列中微生物组与疾病(调整年龄组后)和年龄组(调整疾病状态后)的变化。特定队列的分析确保观察到的变化不是由于特定国家地区微生物组组成的差异造成的。然而,在每个队列内部,不同年龄组的疾病和对照样本的代表性存在偏差(如表1所见)。此外,在八个队列中的四个队列中,对照和疾病个体的年龄变化存在显著差异,如图D中的beanplots所示。
接下来,我们使用了两种不同的方法来探索年龄对疾病-微生物组特征的影响。在第一次分析中,在每个特定疾病的国家级别队列内,我们进行了PERMANOVA分析,研究了疾病特征与年龄组之间的交互作用的影响,同时调整了国家的影响(鉴于不同疾病队列内有来自不同国家的样本)以及疾病和年龄组的独立影响(表2)。两个元数据类别之间的交互作用衡量了微生物组相对于一个元数据(在本例中是疾病)的变化受另一个元数据(即年龄组)变化影响的程度。对于三种疾病(IBD、CRC和息肉),疾病与年龄组的交互作用的影响是显著的(所有p<0.05;调整上述混杂因素后的PERMANOVA)。对于T2D的影响也几乎是显著的(p<0.08;PERMANOVA)。这表明,即使在考虑了地区变异和疾病与年龄的个体影响之后,对于这些疾病,与疾病相关的微生物组变化显著受到个体年龄组变化的影响。对这些影响的进一步研究(通过R2测量)也表明,虽然IBD和CRC作为疾病特征的协变量,年龄的影响最大,而T2D和息肉相对较低但仍然显著(表2)。

表2. PERMANOVA分析结果,研究了疾病特征与年龄组之间的交互作用的影响,调整了国家(在大陆队列内)的影响以及疾病和年龄组的独立影响。
在第二次分析中,我们使用了随机森林(RF)分类器模型。随机森林模型属于基于集成的机器学习方法(基于决策树),可以用来根据一系列预测特征预测给定的特征(在本例中是疾病状态)。随机森林不仅可以用来判断预测因子与响应之间的关联强度(基于预测的准确性或曲线下面积(AUC)),而且还可以用来判断在一个观察集上识别的关联是否可以外推到另一个观察集。因此,随机森林模型在基于微生物组的研究中被常规使用,不仅用来量化和表征微生物组与各种疾病的关联(Feng等人,2015年;Karlsson等人,2013年;Pasolli等人,2016年;Qin等人,2012年),而且还用来研究在一组个体中预测的关联在另一组个体中的可转移性(He等人,2018年;Thomas等人,2019年)。
我们特别关注特定疾病的国家级别分组(以尽可能确保区域同质性)。随后,我们将受试者(对应于每种疾病的国家级别队列)分为三个年龄组(如上所述),并进行了100次迭代,每次在属于一个年龄组的样本子集上训练随机森林分类器,并在来自同一年龄组的样本上评估疾病分类性能(排除用于训练的样本)(同年龄组分类)或其他年龄组的样本(不同年龄组分类)(同时保持训练和测试数据集大小在不同年龄组之间恒定)(详见图2—图补充1和材料方法了解详情)。
总之,在每个疾病-年龄组情景中,我们使用了两种不同的方法来评估同年龄组分类和不同年龄组分类之间的疾病分类性能是否有显著差异。在第一种方法中,对于100个迭代重采样的分类器模型,我们使用配对Wilcoxon符号秩检验比较了同年龄组分类得到的AUC与不同年龄组分类得到的AUC。在第二种方法中,使用置换检验,对于100个迭代重采样的分类器模型,我们比较了年龄组队列的实际AUC差异(同年龄组分类 - 不同年龄组分类)是否与随机情况下预期的差异有显著不同(通过置换受试者的年龄组标签生成的空模型分布)(图2—图补充1和材料方法)。
数据显示不同年龄组的各种疾病之间存在巨大差异。具体来说,在13种疾病-年龄组情景中的10个(涵盖五种疾病)中,与在不同年龄组进行测试相比,在同一年龄组训练和测试的分类器在疾病预测AUC上有显著更高的结果(图2),分类性能的提高显著高于随机机会预期的水平(通过基于置换检验策略获得)(图2—图补充2)。这证实了微生物组-疾病关联具有以年龄为中心的趋势。此外,差异的程度在IBD、CRC和T2D中表现得尤为明显(上述趋势在所有年龄组中都有所体现),反映了PERMANOVA分析中观察到的模式(表2)。CRC和息肉的特征在于明显的类似年龄特异性趋势,其中老年年龄组的分类AUC显著下降(即在老年年龄组训练或测试的分类器的AUC较低),表明这些疾病在老年年龄组的微生物组-疾病特征较弱(图2)。对于这两种疾病,在同年龄组和不同年龄组疾病分类之间的AUC差异在老年人中也有所减少(在息肉的情况下,这种差异与随机机会预期的差异没有显著不同)(图2—图补充2)。
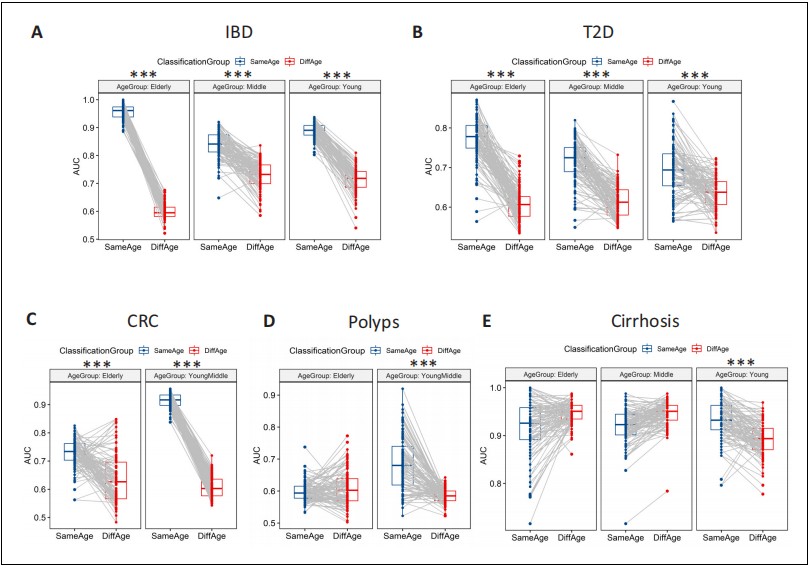
图2. 微生物组-疾病特征显示特定年龄组中心趋势。箱形图展示了当在一个年龄组上训练的分类器在相同(标记为SameAge或同年龄组分类)或不同年龄组(标记为DiffAge或不同年龄组分类)上进行测试时,疾病分类下曲线面积(AUCs)的变化情况,针对(A) IBD (B) T2D (C) CRC (D) 息肉和(E) 肝硬化。每个点表示使用每个基于100个子样本的随机森林分类器模型在测试来自同年龄组(蓝色)或不同年龄组(红色)样本时获得的中位数AUC(20次迭代)。同年龄组和不同年龄组分类的中位数AUC值通过灰色线连接。在同年龄组分类与不同年龄组相比,分类AUC有显著增加的情况被标示出来(使用显著性的P值)。经过Holm方法校正后的Wilcoxon符号秩检验显著性P值标示为***: p<0.001, **: p<0.01, *: p<0.05。
接下来,我们寻求识别与年龄相关的疾病标记物的差异,以及这些标记物与当前已知的微生物组-疾病关联之间的对应关系。除了量化微生物组特征的强度和可外推性之外,迭代的随机森林模型还提供了贡献于模型的分类群列表以及它们的相对重要性得分(针对每种疾病年龄组情景)。因此,在第一步中,基于每种疾病的RF分析(如上所述),我们计算了每个分类群的年龄组特定的特征重要性得分,然后为每个年龄组单独筛选出具有前85百分位标记得分的分类群(见材料和方法;图3—图补充1;图3—源数据1)。比较不同年龄组的前85百分位标记物显示,受年龄影响的标记物(或至少在一个年龄组中有差异关联的标记物)的比例从CRC的30%到IBD的50%不等(其他三个超过35%)(图3—源数据1;图3—图补充2)。然后,使用迭代RF模型获得的特征重要性得分在不同年龄组之间进行了比较,具有显著差异的特征重要性得分的分类群(跨年龄组)(IBD、T2D和肝硬化的Benjamini FDR < 0.01 Kruskal-Wallis检验,CRC和息肉的Mann-Whitney检验)随后被识别出来(图3—源数据1)。
这些结果表明,微生物组与疾病的关联既有年龄组特定的部分,也有年龄组独立的成分。然而,许多年龄组特定的微生物组-疾病关联的变化可能也反映了伴随一般衰老的变化。年龄是影响微生物组组成的第二大因素,因此在去除了衰老影响后,调查这些年龄特定的疾病-微生物组关联的变化是很重要的。其次,虽然将样本分入国家级别的箱子确保了比较组之间的足够地区同质性,但在不同疾病中,特定年龄组的某些地区代表性仍然存在偏差(图1—图补充3)。鉴于RF模型无法内在地调整这些混淆效应,我们使用基于线性回归的策略对此进行了探究,该策略比较了年龄组对分类群疾病关联模式的影响程度(疾病:年龄组)与疾病和年龄组个别影响相比(见材料和方法;图3—图补充3)。
我们特别研究了那些在特征重要性得分上有显著差异的分类群(图3—源数据1),并识别了那些在疾病与年龄组交互作用下具有显著更高影响的分类群,而不是仅年龄组本身(Log似然检验单侧p<0.05)(图3—源数据2;图3)作为最终验证的“严格年龄特定疾病标记物”的列表。值得注意的是,在那些在特征重要性得分上有显著差异的分类群中,也在线性回归方法中得到验证的分类群的百分比,显示出特定于疾病的趋向,反映了我们在基于PERMANOVA(表2)和基于RF的分析(图2)中观察到的。具体来说,对于IBD和CRC,超过63%的在年龄组间标记得分上有显著差异的特征也在基于线性回归的验证中得到了验证,这表明对于这些疾病,即使在考虑了衰老的整体效应后,大多数年龄组特定的疾病关联仍然存在。相比之下,对于息肉和肝硬化,这些重叠显示出逐渐减少的趋势(图3—图补充4A)。接下来,我们将在这里识别的年龄相关的疾病标记物与最初报告的疾病标记物进行了比较。对于四种疾病(即不是CRC),我们从每项原始研究中编制了一份已知的与疾病相关的标记物列表(Feng等人,2015年;Franzosa等人,2019年;Karlsson等人,2013年;Qin等人,2012年;Qin等人,2014年)。对于CRC,最近的一项研究对所有主要的队列进行了荟萃分析(包括在curateMetagenomicData中的那些)(Feng等人,2015年;Vogtmann等人,2016年;Zeller等人,2014年),以及三个新测序的队列(Thomas等人,2019年),以产生一套经过跨队列验证的精炼的CRC标记物。
我们利用这个列表来编制已知的CRC标记物集。将这些关联的列表与原始研究报告的所有已知标记关联的预编译列表进行比较(图3—源数据3)(Feng等人,2015年;Franzosa等人,2019年;Karlsson等人,2013年;Qin等人,2012年;Qin等人,2014年;Vogtmann等人,2016年;Zeller等人,2014年),发现在息肉的原始研究中识别的分类群中有40%,在CRC和IBD中有超过63%,在T2D中有58%,在肝硬化中有25%,在年龄组之间显示出显著的特征重要性得分差异,从而强调了年龄相关的疾病-微生物组关联在疾病中的普遍性(图3—图补充4B)。即使在经过衰老效应去卷积的“严格年龄特异性标记物”的最终列表中,重叠率也从T2D和息肉的20%到CRC的高达63%不等(图3;图3—图补充4C)。对于IBD的效果尤为明显,许多已知的标记物(属于Lachnospiraceae、Escherichia、Clostridium clostridioforme、Clostridium nexile、Blautia producta的物种)在某些年龄组中甚至没有被识别为前85百分位(图3)。对于CRC,一些众所周知的标记物Fusobacterium nucleatum、Parvimonas micra、Peptostreptococcus stomatis和Porphyromonas asaccharolytica,尽管在不同年龄组中被识别为前85百分位,但根据RF特征重要性得分以及线性建模的结果,显示出年龄特定的趋势。对于T2D的一些标志性分类群(Faecalibacterium prausnitzii、Roseburia intestinalis、Haemophilus parainfluenzae、Bacteroides intestinalis和Alistipes indistinctus)和息肉(Bacteroides dorei)也观察到了同样的模式。当我们比较每个标记物在不同年龄组中被识别为大于85百分位得分的百分比(在每个年龄组的100次迭代中)(图3—图补充4D)时,这些关联的强度和稳定性的变化进一步明显。一些已知的CRC(Parvimonas micra、Eubacterium eligens)和T2D(Bacteroides intestinalis、Haemophilus parainfluenzae)的标记物在某些年龄组中超过80%的迭代中被识别为前85百分位,但在其他年龄组中不到50%。这对微生物组导向的诊断/治疗策略在不同年龄组的有效性有影响。

图3. 特定分类群显示出与年龄组相关的疾病关联趋势。热图显示了与所示疾病在不同年龄组(Y:年轻;M:中年;E:老年)有差异关联的分类群的标记得分列表。对于每种疾病,这个物种列表被选定为那些至少在一个年龄组中位于前85百分位特征之一,并且在至少两个年龄组中显示出特征重要性得分的显著变化的物种。这些分类群进一步通过线性回归方法进行了验证,以确保在考虑与衰老相关的独立变化后,它们与疾病特定年龄组的关联仍然显著。物种的字体颜色表明这些物种是否在原始研究中被报告与给定疾病有关联(深蓝色:之前有关联;黑色:未关联)。对于每种疾病,热图旁边的热图(紧挨着相应热图的右侧)显示了在每个年龄组中被识别为前85百分位标记的不同分类群(以蓝色表示)。
接下来,我们测试了观察到的疾病关联的差异趋势是否受到特定队列的影响。对于CRC(结直肠癌),我们观察到年龄组对疾病特征的影响最为显著,这些特征影响了超过63%已知的CRC标记,并且即使在考虑了国家特定的表现和去除了衰老影响后也得到了验证。此外,三个新测序的队列(称为“队列1”、“队列2”和“WirbelJ_2019”,作为一个整体称为“验证队列”)具有不同年龄组样本的充分代表性(补充文件1)(Thomas等人,2019年;Wirbel等人,2019年),并且代表了最初在鉴定差异相关标记时未包含的新数据。在整理的MetagenomicData存储库中,CRC疾病样本来自三个队列,即ZellerG_2014、VogtmannE_2016和FengQ_2015(Feng等人,2015年;Vogtmann等人,2016年;Zeller等人,2014年)。为了探索这些关联的可复制性(不考虑衰老的一般影响)以及理解这些差异关联的生物学基础,我们在验证队列上验证了在这个队列上设计的具有年龄意识的RF分类模型(称为训练集1)(图4A;上层面板)(使用与图2—图补充1中描述的类似策略)。疾病分类的年龄特异性趋势与之前获得的相似(图2),即在相同年龄组上训练的分类器在测试时具有最高的分类性能,而在测试老年人时,分类AUC显著降低。相比之下,对于在老年人上训练或测试的分类器,性能显著下降(在同年龄组和不同年龄组测试之间的差异不显著)(图4A;图4—图补充1)。
我们进一步通过从“验证队列”中取样本子集(称为训练集2)并在非训练子集上进行测试,生成了类似的迭代年龄组特异性微生物组疾病预测模型。分类模式保持不变(图4A;下层面板与上层面板比较)。这些结果表明,年龄组特异性微生物组-疾病特征的变化是稳定的,并非来源于训练数据集的偏差。 然后,我们检查了已知CRC标记的年龄依赖关联在两个训练模型之间的重叠(图4B)。在19个跨队列相关的CRC相关分类群中(Thomas等人,2019年),13个(67%)在训练集1中与任一年龄组有差异关联(经过FDR校正的p值<0.1)。其中9个(77%)在训练集2中可以复制相同的方向性(图4B)。有趣的是,在最近的元分析报告的顶级CRC预测特征中,有六个只在年轻/中年类别中与更高的预测能力相关(图4B)。只有一个与老年微生物组相关联。 接下来,我们检查了CRC在年轻/中年和老年年龄组之间的特征关联在不同队列中的稳定性。
本研究采用的自助法方法使得能够为疾病和对照受试者的多个子集获得微生物组-疾病特征。在微生物组特征强大且稳定的情况下,分类器每次迭代得到的微生物组-疾病关联(在这种情况下是每个分类群的特征排名概况)应该是相似的(跨队列)。然而,在这个特定的情况下,虽然两个队列中年轻/中年个体的分类器概况相对相似,但老年特异性疾病分类器获得的队列间距离显著更高(表明疾病特征变化和可复制性丧失)(p<2.2e-16;Mann Whitney Test)(图4C)。
尽管在比较已知CRC特异性标记的丰度时没有观察到特定趋势,但前十个标记中的五个(Fusobacterium nucleatum、Peptostreptococcus stomatis、Gemella morbillorum、Parvimonas micra和Parvimonas spp)在老年对照组中的患病率显著更高(在两个数据集中都可以复制)(图4D)。这导致它们在老年年龄组中(对照和患病个体之间的差异)的效应大小显著减少。因此,与衰老相关的改变可能使老年人容易患上与微生物组相关的特定疾病,如CRC。
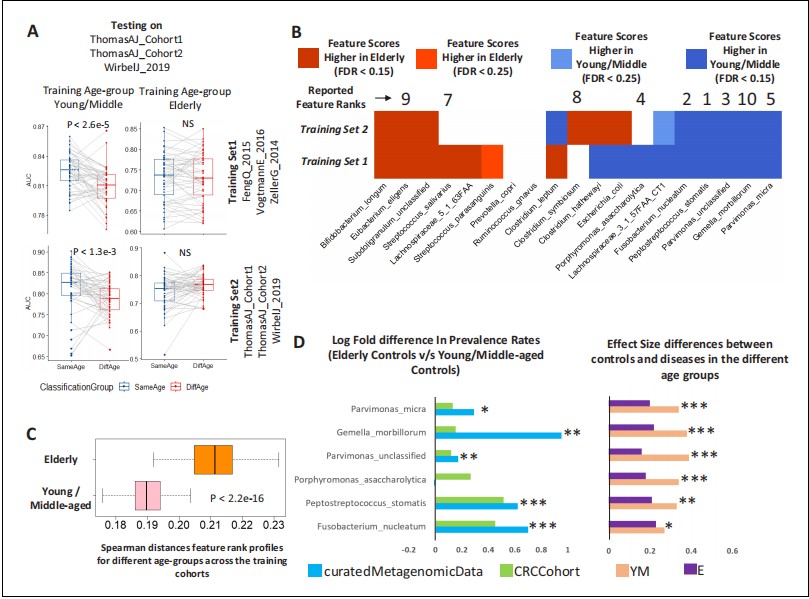
图4. 年龄依赖的CRC特异性标记在多个队列中可复制,与衰老相关的改变使老年肠道微生物组呈现疾病样特征。(A)上层面板的箱形图显示了在三个队列(Training_Set1: ZellerG_2014、FengQ_2015和VogtmannE_2016)中训练的不同年龄组(YM: 年轻/中年;E: 老年)的分类器在三个验证队列(ThomasAJ_Cohort1、ThomasAJ_Cohort2和WirbelJ_2019)的数据集上测试时获得的AUC值分布。下层面板显示了同样的情况,但是使用了在验证队列内训练的年龄组特异性分类器(Training_Set2)。两种分类模型生成了相同的分类趋势,表明疾病特征的年龄组特异性可复制性。点颜色的描述与图2相同。 (B) 两个独立队列中已知CRC标记的年龄组依赖关联,即Training_Set1(整理的MetagenomicData)和Training_Set2(验证队列)。蓝色阴影表示年轻/中年中特征重要性得分更高,红色表示老年中特征重要性得分更高。FDR p<0.15表示在老年或年轻/中年中被识别为高特征的重要性得分,经过Benjamini-Hochberg校正的Mann-Whitney测试p值<0.15。FDR p<0.25表示在老年或年轻/中年中被识别为高特征的重要性得分,Mann-Whitney测试p值<0.25。在19个已知并经过验证的CRC标记中(来自Thomas等人,2019年),有13个在两个年龄组中的MetagenomicData队列显示出显著的特征重要性得分差异。对于这13个标记中的9个,关联模式可以在验证队列中复制,进一步表明获得结果的可复制性。Thomas等人(2019年)获得的前10个标记的特征排名也显示在这里。前10个标记中有6个显示出增加的关联性,但仅在年轻/中年中。只有一个标记与老年人相关联。这表明老年人失去了疾病特征。 (C) 不同年龄组训练的疾病分类器获得的特征排名概况的跨队列Spearman距离(详见材料和方法)。稳定的疾病特征会导致跨队列的可复制物种排名概况,从而产生较低的Spearman距离。虽然年轻/中年的情况是这样,但不同队列中为老年人获得的标记显示出显著高的Spearman距离(显示出显著的变化和缺乏疾病特征)。 (D) 老年对照组与年轻/中年对照组相比,前六个与CRC相关的标记的患病率的对数比率(在整理的MetagenomicData和CRC特异性队列中都是如此)。正值表示老年对照组患病率更高。增加的显著性也指出了(使用Fisher方法结合的fishers'精确测试的p值)作为***: p<0.001, **: p<0.01, *: p<0.05。老年人的增加特征是老年对照组与患病个体之间的效应大小差异显著减少,导致隐藏的标记。
接下来,我们调查了疾病是否具有不同的微生物分类群增加或减少的模式,即使是在不同的年龄组中也是如此。我们首先关注单独的研究队列(图1C)。对于每种疾病-年龄组情况,我们通过比较(研究匹配的)对照和患病样本中的丰度趋势(Mann Whitney 测试 p<0.05),来确定相应顶级疾病预测因子的关联方向性(疾病中增加与减少)。对于五种疾病中的四种(即除了息肉),微生物组改变的方向性发生了显著变化,其特征是随着年龄增长,获得的特征数量显著减少(Fishers’精确测试 p<0.05;图5—图补充1)。为了增加统计效力并检查上述趋势是否在更大的队列中保持,我们使用“大陆匹配”的疾病和对照进行了分析(使用从Mann Whitney测试获得的相当严格的Benjamini-Hochberg校正P值<0.1的阈值)。趋势仍然相似。对于老年人,微生物组改变(除肝硬化和息肉外的所有疾病)的特征是失去的分类群数量显著增加(图5A)。
具有显著疾病关联方向性的疾病特异性标记列表呈现在图5—源数据1中。因此,这些疾病中的大多数微生物组改变特征是逐渐从由获得的微生物组成分主导的状态转变为老年人中控制相关分类群的增加丧失。 这些发现表明,随着年龄的增长,特定疾病中的微生物组组成发生了显著变化。在年轻和中年个体中,微生物组的改变可能更多地表现为与疾病相关的分类群的增加。然而,在老年人中,情况似乎发生了逆转,与疾病相关的微生物组改变更多地表现为与健康对照相比的分类群的丧失。这种从获得到丧失的转变可能反映了与年龄相关的微生物组组成的动态变化,这些变化可能与疾病风险和进展有关。此外,这种年龄依赖的微生物组响应的共同模式可能表明,尽管不同疾病具有独特的微生物组特征,但它们都可能受到相似的生物学过程的影响,这些过程与衰老和疾病易感性有关。
因此,这些结果强调了在疾病研究中考虑年龄因素的重要性,并为进一步探索微生物组与特定疾病之间的关系提供了新的视角。 在一项研究报告中,不同疾病间改变的分类群的重叠为51%(Duvallet等人,2017年),而另一项研究报告称,通过聚合多项研究中的疾病样本来训练通用的疾病对照分类器,仍然能够以AUC大于0.8的准确度区分对照组和疾病组(Pasolli等人,2016年)。鉴于微生物组成随着年龄增长而变化,我们研究了年龄对这些共享疾病响应程度的影响。我们设计了通用的疾病分类器(使用随机森林),取对照组和疾病个体的等量子样本(包含每种疾病相同数量的样本,以防止在分类性能中出现疾病/年龄组特定的偏差)(详见材料和方法以及图1—图补充3)。
虽然这些通用疾病预测模型在年轻/中年人群中的表现很高(中位AUC: 0.79),并且与早期研究报道的相似(Pasolli等人,2016年),但同样的模型应用于老年受试者的数据时,其性能显著降低(p<1e-7)(图5B)。此外,在这些模型中,虽然在疾病预测敏感性方面没有观察到显著差异(p<0.13),但预测的特异性(即识别健康个体的准确性)对老年人来说显著降低。这不是由于不同疾病样本的不同代表性造成的,因为我们确保了所有年龄组中所有疾病的平等代表性。因此,与以前的元分析相比,老年受试者之间的共享疾病响应显著降低,主要是在区分非疾病个体方面。此外,我们对年龄对疾病相关分类群影响的澄清(图3—源数据1;图3—源数据2;图5—源数据1)提供了一套改进的特征集,用于这些疾病的基于微生物组的诊断。随着年龄的增长,微生物组成模式的这种独立于疾病的改变,以及通用疾病分类器无法区分非疾病对照的情况,是令人感兴趣的,可能是由于肠道微生物组中有益细菌的流失造成的,这反过来可能受到饮食和药物等多种因素的影响。这种日益增加的流失可能导致以更高的个体间变异性、病原生物的增加丰富度(导致疾病特征的丧失)为特征的失调性配置,从而使微生物组更容易受到疾病的影响。
因此,我们接下来使用年龄组内的Spearman距离,检查老年对照组的样本是否与年轻/中年对照组相比,在不同大陆地区具有显著更高的变异性(图5—图补充2)。与我们的假设一致,对于欧洲和北美,老年个体的肠道微生物组成与年轻和中年对照组相比显著更加可变。然而,在亚洲(中国)队列中没有观察到这一点。有趣的是,肝硬化组既不显示与疾病特征的关联,也只包含了来自亚洲的受试者。 接下来,我们试图表征共享疾病响应的元素。值得注意的是,对关联方向性的仔细检查表明,特定的分类群与多种疾病呈现出一致的关联趋势(基于图5—源数据1中显示的趋势)。分类群增加或减少的总体模式包括了早期研究观察到的几种趋势(Duvallet等人,2017年;Pasolli等人,2016年)。例如,Streptococcus anginosus和Fusobacterium nucleatum被检测出在多种疾病中增加。同样,属于Roseburia spp.(R. hominis)的物种被检测出减少。我们共识别出61个分类群,在年轻/中年或老年人群中显示出与多种疾病一致的关联方向性(图5C)。根据它们在不同年龄组中共享响应的差异检测特征,我们将这些分类群分为六个不同的组,即G1(在所有年龄组中疾病状态增加)、G2(仅在老年人中疾病状态增加)、G3(仅在年轻/中年人中疾病状态增加)、L1(在两个年龄组中疾病状态都减少)、L2(仅在老年人中疾病状态减少)和L3(仅在年轻/中年人中疾病状态减少)(图5C)。许多以前报道与多种疾病中共享增加或减少相关的物种(Duvallet等人,2017年;Pasolli等人,2016年)属于G3组,即它们在年轻和中年人群中显示出增加或减少的类似趋势(如早期报道的),但在老年人中并非如此。这些包括链球菌、Fusobacterium nucleatum、大肠杆菌和脆弱拟杆菌。
相比之下,另一组物种,包括Ruminococcus torques、Solobacterium moorei和Streptococcus mitis,仅在老年人中与多种疾病相关。最后,我们识别出一个独特的物种组(G1),在老年和年轻/中年人群中跨疾病增加。这些包括一群梭菌(C. bolteae、C. symbiosum、C. hathewayi、C. citronae、C. asparagiforme)(图5C;有关个别疾病的详细信息,请参见图5—源数据1)。这些分类群已在不同疾病和/或类病状态(T2D、息肉、CRC和自闭症)的独立研究中被识别出来(Pequegnat等人,2013年;Qin等人,2012年;Sinha等人,2019年;Yu等人,2017年),但在这里首次显示,它们是跨疾病共享增加响应的一部分。基于这些发现,我们假设这个特定的G1组分类群构成了一个与受影响个体的一般病理生理衰竭相关的共享疾病响应。
这些发现对于理解微生物组与疾病之间的关系具有重要意义。通过识别出与多种疾病相关的共享微生物组响应,我们可以更好地理解疾病的共同生物学基础,并可能为开发新的诊断和治疗方法提供线索。特别是G1组分类群的发现,表明了一种可能的跨疾病的通用病理生理机制,这可能与老年和年轻/中年人群中的病理生理衰竭有关。这种共享响应的识别为未来的研究提供了一个有价值的框架,可以用来探索微生物组如何与宿主的病理生理过程相互作用,以及这些相互作用如何影响疾病的发展和进展。此外,这些信息可能有助于识别新的生物标志物,这些生物标志物可以用于早期诊断、疾病风险评估以及监测治疗反应。
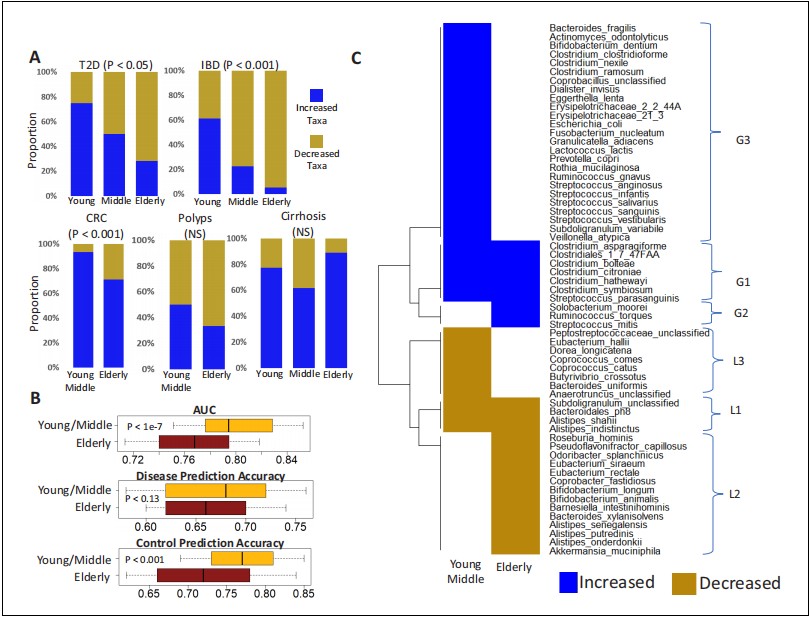
图5. 与年龄相关的微生物组变化影响特定疾病的分类群丰度变化,以及多种疾病共有的微生物组响应。(A) 对比五个疾病在年轻、中年和老年人群中介于丰富和稀少的疾病特异性标记分类群的相对比例。对于每种疾病-年龄组情况,我们通过比较特定年龄组的对照和患病样本中的丰度趋势,检查相应顶级疾病预测因子的关联方向性(疾病中丰度增加与疾病中丰度减少)(详见材料和方法)。为确保获得的结果不受微生物组组成区域变化的影响,我们再次将这些比较限制在疾病特异性大陆队列中。(B) 比较老年组和年轻/中年组获得的通用疾病预测模型的疾病预测AUC、疾病分类敏感性和对照分类特异性。总体而言,通用疾病分类器在老年人群中的性能显著下降,表明共享的微生物组响应在老年人中可能减少。此外,性能损失尤其在区分疾病对照样本方面显著。(C) 热图显示在老年和年轻/中年人群中至少两种疾病中一致增加或减少趋势的标记物种。蓝色表示在两种或更多疾病中一致增加,红色表示在两种或更多疾病中减少。根据它们在两个年龄组中的增加或减少模式,这些分类群可以分为六组,即G1-G3和L1-L3。
这张图展示了年龄如何影响特定疾病和多种疾病共有的微生物组变化。通过比较不同年龄组中疾病相关分类群的丰度变化,我们可以更深入地理解微生物组与疾病之间的复杂关系,以及年龄如何影响这些关系。这些发现强调了在疾病研究和临床实践中考虑年龄因素的重要性,并可能有助于开发针对不同年龄段人群的更有效的诊断和治疗方法。此外,通过识别在老年人和年轻/中年人群中一致变化的分类群,我们可以更好地理解这些变化背后的生物学机制,并可能识别出新的生物标志物或治疗靶点。
老年个体的脆弱性特征是多个系统功能的降低。我们研究了是否可以在ELDERMET队列中验证分类群与脆弱性的关联(补充文件2),对于这个队列,我们既有随机宏基因组数据也有粪便代谢组数据。使用随机森林回归(带五折交叉验证),我们可以根据肠道微生物组的分类组成预测个体的脆弱性(测试社区和居住护理受试者)(R = 0.79;图6A;图6—图补充1)。然后,我们计算了图5C中六个增/减通用疾病响应组在脆弱性预测模型中的平均特征重要性排名。验证我们之前的观察,G1组分类群在脆弱性(功能独立性测量(FIM))预测中拥有最高的平均特征重要性排名,表明这个组在ELDERMET受试者中对脆弱性具有最高的预测能力(图6B),其次是G2组(仅在老年人中跨多种疾病增加)。接着是我们早期关于老年人特有的共享微生物组响应标记的发现,这是由L1组跟随的。最佳的八个分类群具有最高的脆弱性预测能力(图5C;图6C;图6—图补充1)。所有这些分类群在脆弱性中都更丰富(与FIM负相关,与脆弱性正相关)(图6—源数据1A–B)。八个分类群中的五个属于G1组(图6C;图5C)。
因此,这些在ELDERMET队列中独立验证的发现,确认了我们早期对特定老年相关的通用疾病响应组的识别(图5C)。这些发现进一步强调了微生物组组成与老年个体脆弱性之间的密切关系,并且指出了G1组分类群在老年人群中可能作为一个潜在的生物标志物或治疗靶点的重要性。通过这种类型的研究,我们可以更好地理解老年相关疾病和脆弱性的微生物组基础,并可能有助于开发新的预防和干预策略,以改善老年人的生活质量和健康。
如果微生物群的改变有助于疾病发生,微生物代谢产物可能是效应分子。当前研究的一个显著特点是它基于宏基因组数据,不同于以往的基于16S的微生物组-疾病元分析(Duvallet等人,2017年)。这使我们能够在更细的分类群水平上获得分类组成,进而我们可以利用组成分类群的实验验证功能谱来预测微生物组的代谢物谱。为此,我们使用了虚拟人类代谢数据库(Noronha等人,2018年),以及一种搜索实验验证的功能谱的方法,这些功能谱代表了不同肠道相关物种中不同代谢物的生产和消费模式(Sung等人,2017年)。因此,我们可以从992个物种中整理出300多个代谢物谱(消费/生产/降解)(图6—源数据2)。
共有82个代谢物谱与FIM评分有显著关联(Spearman Rho;FDR小于0.25)(图6—图补充2)。我们观察到这种代谢物谱的关联分析反映了与特定化合物的生物可利用性相关的脆弱性改变,其中许多之前已经显示出与健康相应的关联,从而验证了我们的发现(Claesson等人,2012年)。具体来说,脆弱性的发作与肠道细菌消费SCFA的增加有关(伴随着SCFA丁酸的生产同时减少),有益氨基酸色氨酸的消费增加,以及与T2D相关的支链氨基酸苏氨酸的生产增加。
我们首先关注了脆弱性标记分类群,并鉴定了13个与八个脆弱性标记分类群显著相关的代谢物谱(图6D)(详见材料和方法)。这13个代谢物谱的子集本身就能以0.60的R值预测脆弱性(实际和预测的FIM值之间),这比从代谢物谱中移除这些特征后获得的R值显著更高(图6D;从左数第二的顶部面板)。第一组关键代谢物谱包括主要胆汁酸(即胆酸(CA)和陈脱氧胆酸(CDCA))的降解,产生疏水性次级胆汁酸(鹅去氧胆酸:LA;脱氧胆酸:DCA)。
我们对ELDERMET受试者的粪便代谢组谱进行了实验测量,验证了这一预测的代谢功能,其中脆弱个体的粪便代谢组以LCA(及其衍生物)和DCA的显著更高水平以及CA和CDCA的显著更低水平为特征,与非脆弱个体相比(图6D)。与此功能相关的两个物种,即Clostridium scindens和Clostridium leptum,既不属于G1组也不属于G2组(与本研究中它们的脆弱性关联相反)。这些物种先前分别与Clostridium difficile抵抗和IBD有关(Buffie等人,2015年;Manichanh等人,2006年)。与它们的治疗相关性相反,这些物种的这一特定功能,即产生更高水平的疏水性胆汁酸,也与CRC和非酒精性脂肪肝病(NAFLD)的发作有关(Tsuei等人,2014年)。
最近对CRC肠道微生物组的一项元分析也预测,次级胆汁酸生产的增加是关键的CRC特异性功能特征(Wirbel等人,2019年)。 与粪便代谢组分析中识别的虚弱特异性标记相关的另外两种代谢物功能也得到了验证,它们是p-甲酚的产生(测量的p-甲酚与FIM呈负相关:p<0.01;与社区和长期居住者的FIM相关系数R=0.29;仅长期居住者为R=0.47)和乙醇的产生(与非虚弱个体相比,长期居住队列中虚弱个体的粪便代谢组中显著更高水平:p<0.02)。虽然p-甲酚是一种由特定肠道细菌物种(包括艰难梭菌)产生的细胞毒素,与微生物群的改变、大肠癌和胰岛素抵抗有关(Fau等人,1976年;Khan等人,2014年),但肠道中高水平的乙醇产生不仅与非酒精性脂肪肝病(NAFLD)、动脉粥样硬化和SIBO的发生有关,而且还与炎症标志物水平的增加有关(Elshaghabee等人,2016年)。
同样,丙酮和氨(NH3)的产生与细胞毒性增加、小肠细菌过度生长以及胰岛素抵抗有关(Baskaran等人,1989年;Ghoshal等人,2017年;Khan等人,2014年)。尽管实验中心微生物组样本没有粪便代谢组数据,但使用humann2获得的特定微生物途径的丰度与这些代谢物的产生有关,即丙二醇到丙酮,以及尿囊素到氨/二氧化碳/乙醛酸的转化,预测在虚弱个体的微生物组中显著更高(图6D;下方板)。与虚弱标记相关的另一种功能是胆碱消耗和三甲胺(TMA)的产生。特定的肠道微生物物种,包括柠檬酸梭菌、梭状芽孢杆菌和哈瑟韦氏梭菌,将胆碱降解为三甲胺,这与动脉粥样硬化有关(Martı´nez-del Campo等人,2015年)。通过测序深度标准化的特定细菌CutC酶的丰度分析,该酶催化这一转化,在虚弱个体的肠道微生物组中显示出显著更高的代表性(图6D;右上角板)。这种与结直肠癌(CRC)的关联也在当前研究使用的元分析数据集中报告过(Thomas等人,2019年)。
因此,从表面上看,这组微生物组标记物可能在功能上推动宿主进入越来越容易受到各种疾病影响的状态,也加速了虚弱的发作。有趣的是,发现G1-G3标记物中类似的代谢物关联表明,虚弱相关标记物的代谢物产生特征与G3组的分类群(特别是乙醇和NH3的产生)有重叠,表明在一般微生物群破坏状态下富集的不同分类群,无论它们的组成差异如何,都包含某些可能将肠道生态系统推向对宿主有害状态的代谢特征(图6—图补充3)。同样,将G1-G3组的分类群的代谢特征与不同组的丢失分类群L1-L3的代谢特征进行比较,也揭示了一个有趣的模式,即“获得”组与简单糖(乳糖、松三糖、木糖、蔗糖、拉菲糖)的消耗不成比例地相关。相反,“丢失”组则富集于膳食益生元的消耗,如木寡糖、果寡糖、菊粉和产生丁酸和琥珀酸。
最近对迁移到美国的泰国个体的肠道微生物组的研究发现,随着停留时间的增加,与降解膳食纤维(如木聚糖、阿拉伯木聚糖、纤维二糖、普鲁兰、葡甘露聚糖和抗性淀粉)相关的代谢功能丧失,导致微生物组功能的更广泛丧失(Vangay等人,2018年)。这些模式反映了特定微生物群的代谢能力,对于基于饮食的微生物组恢复策略在老年人中尤其重要,与年轻人和中年人相比,控制相关分类群的丧失对疾病的影响更大。

图6. 虚弱相关标记在两个年龄组的多种疾病中具有共享的正向关联,并具有特定的代谢特征。 (A) 老年个体(生活在社区或养老院,即长期居住者)的ELDERMET队列中,实际的功能独立性测量(FIM)值与通过随机森林对微生物组特征预测的FIM值的对比。 (B) 在ELDERMET队列中,用于预测FIM(虚弱的反向测量)的各种分类群(在图3中识别)的平均排名。 (C) 在随机森林模型中,预测FIM的八个具有最高预测能力的标记的变量重要性得分。比较高FIM和低FIM个体之间标记的丰度表明,所有这些标记都与虚弱状态相关。 (D) 中央面板的网络显示与顶级标记显著相关的13个代谢物概况。分类群标记在中心标示。消耗概况在上半部分(粉红色八边形中),生产概况在下半部分(黄色八边形中)。边缘表示存在。
顶部面板从左数第二是使用仅包含(D)中的13个代谢物标记的迭代自助法随机森林模型(20%用于训练,其余80%用于测试)获得的预测FIM值与实际FIM值之间的相关性。上下面板显示了使用测量的代谢物、饮食消耗概况、特定的微生物途径丰度以及使用humann2识别的CutC基因家族丰度(通过箱形图比较虚弱与非虚弱个体之间的概况,或散点图显示测量的代谢物水平与个体FIM值之间的相关性)获得的预测代谢物标记的验证(由箭头指示)。13个代谢物中有11个可以使用这些策略之一进行验证。

通过识别与不同疾病相关的特定年龄的微生物组关联,这项研究有可能为特定年龄群体定制基于微生物组的诊断策略提供信息。在疾病-微生物组分类器中考虑年龄已经阐明了许多疾病共有的微生物组改变,并缩短了与常见非传染性疾病特别相关的分类群的清单。此外,观察到的分类群关联的整体方向性变化(例如,老年人逐渐出现分类群丧失表型)强调了微生物组恢复策略对于老年受试者的疾病改善可能更为关键(相对于基于抗生素的根除方案)。这一点尤其重要,因为观察到随着年龄增长,可能会出现特定的改变,使宿主更容易受到某些与微生物组相关的疾病的侵袭。
上述模式在使用通用疾病分类器进行的我们分析中得到了进一步观察。分类器区分病例和对照组的能力的特定丧失进一步表明,随着年龄的增长,与健康相关的微生物组标记正在丧失。有趣的是,这些发现与我们早期研究的结果相呼应,随着年龄的增长,核心微生物组的减少伴随着病原生物的增加(O’Toole和Jeffery)。最后,可重复识别与多种疾病相关的特定物种标记集及其特定的代谢物概况(糖类消耗和产生一系列对宿主有害的代谢物)表明,获得这一子集的疾病相关分类群可以将代谢状态转变为类似疾病的状态。
在微生物组-疾病研究中,区分相关性与因果关系需要识别可能与疾病稳健相关的分类群,通过在微生物组-疾病关联研究中考虑年龄可以改进这一点。当前分析中的许多特征在原始出版物中没有报告。这可能有几种原因,即与当前研究相比,原始研究中使用的比较分析协议的变异;通过结合多项研究,当前分析具有更强的能力和更稳健的签名;不同年龄组样本数量的研究偏见。
在这方面,一个关键方面是年龄特异性差异关联在地区/种族间的相对稳定性。 在这项研究中,我们使用了随机森林、线性模型以及在不同地区同质性水平上的验证,不仅减少了地区变异对微生物组签名的影响,还解耦了一般衰老对这些年龄特异性疾病标记的影响。然而,特定疾病-年龄组情况下某些地区样本数量的偏见仍然存在。
目前,大多数在curatedMetagenomic数据存储库和其他验证队列中的数据集来自北美、欧洲和中国。然而,未来特别是来自非西方人群的疾病队列的微生物组数据的可用性将进一步阐明疾病分类的年龄特异性趋势。这尤其重要,因为在当前研究中,我们识别了年龄组之间的地区特异性变化,在北美和欧洲的老年对照组的肠道微生物组与地区匹配的年轻/中年对照组相比变异显著更高,而在亚洲(中国)人群中没有观察到这种差异。未来新的疾病特异性霰弹枪微生物组数据集的可用性将进一步使得能够在其他疾病上进行疾病标记的跨队列验证(正如在当前研究中对CRC所做的)。
需要考虑的另一个因素是饮食的影响。许多在疾病中耗尽的分类群也是那些其生长受到复杂碳水化合物消费促进的分类群。不同人群有特定的饮食模式,这些关联需要被纳入未来适当设计的前瞻性研究中。 由于缺乏关于实验中心(ExperimentHub)的受试者/数据的药物摄入信息(除了已知接受了抗生素治疗并被排除的受试者),我们无法控制药物摄入的变量,这可能是一个主要的混杂因素(Forslund等人,2015年)。以前的研究报告了某些疾病标记与药物摄入的关联。例如,链球菌科被证明与质子泵抑制剂(Proton Pump Inhibitors, PPIs)的摄入有关(Jackson等人,2018年)。
另一项最近研究调查了二甲双胍对当前研究中分析的两个2型糖尿病(T2D)队列的肠道微生物组的影响,发现乳酸杆菌(Lactobacillus salivarius)受到二甲双胍的影响,伴随着大肠杆菌(Escherichia)的增加(Forslund等人,2015年)。然而,没有其他G1和G2标记被报告受到影响。FranzosaEA_2018数据集(与curatedMetagenomicData存储库一起添加)包含了免疫抑制剂、类固醇和美沙拉嗪使用情况的药物使用信息(Franzosa等人,2019年)。
对于这三种药物,我们检查了共享疾病标记的丰度是否受到药物摄入的影响(补充文件3)。我们只能识别出肽链球菌科(在美沙拉嗪治疗中减少)、Coprococcus comes(在美沙拉嗪治疗中增加)和Barnesiella intestinihominis(在类固醇治疗中减少)的关联。此外,调整药物摄入并未改变预测ELDERMET队列虚弱的分类群的丰度(已知药物使用数据)。每个顶级虚弱相关标记不仅在考虑所选药物类型的影响后与不同的虚弱度量有显著关联,而且在高或低药物摄入的个体中独立地被识别为虚弱的前15个预测因子(图5—源数据1c)。然而,与这个有限的药物列表的关联缺失并不能完全排除药物部分作为这些差异关联的驱动因素的可能性。因此,对于像2型糖尿病这样的疾病,这里得到的结果需要谨慎对待。未来的队列研究应该记录所有饮食和药物摄入的测量,以便进一步阐明年龄、生活方式、饮食、微生物组和健康之间复杂的相互作用。
另一个可能的混杂因素可能是疾病出现的年龄段。例如,最近的一项研究发现了癌症阶段与微生物组的相互作用(Yachida等人,2019年);然而,我们还从公布的数据中注意到,51%的年轻受试者(年龄小于60岁)表现为III/IV期癌症,而较高比例(71%)的老年受试者表现为I/II期癌症(p<0.01)。不同年龄组中疾病表现的差异也可能影响疾病-微生物组的标记,因此需要在未来的研究中考虑。 最后一个问题是方法论上的,涉及到为每种疾病-年龄组情景生成的迭代随机森林(RF)分类器中的训练样本之间的非独立性。每次迭代生成的分类器可能会共享训练(和/或测试)样本的重叠,从而可能导致AUC分布范围变窄,这可能会夸大显著性值。然而,差异的大小以及使用多种方法和研究队列所获得结果的一致性清楚地表明了本研究结果的可靠性。
本文译自:Tarini S Ghosh, Mrinmoy Das , Ian B Jeffery et al. Adjusting for age improves identification of gut microbiome alterations in multiple diseases. Elife 2020 Mar 11:9:e50240. doi: 10.7554/eLife.50240.
发表杂志:Elife
影响因子:7.70
通讯作者:Paul W O’Toole
作者单位:University College Cork, Ireland
