


肠道微生物群可能影响人类健康和疾病。尽管在中国人群中已经广泛研究了与疾病相关的微生物群落改变,但仍然缺乏一个全国性的中国肠道微生物群基线。在这里,我们对来自2678名健康中国个体的粪便样本进行了16S rRNA基因测序,这些个体属于八个民族群体,并居住在28个省份的63个县/市。我们鉴定了四种肠道类型,其中三种分别富含普雷沃特氏菌、拟杆菌和大肠杆菌,而第四种没有优势属。通过评估肠道微生物群与地理、人口统计学、饮食、城市化、生活方式和采样月份等六个类别的20个变量之间的关联,我们揭示了地理因素解释了最大的微生物群变异,并阐明了与主食类型、民族和城乡居住的关联模式的差异。具体来说,来自同一地点的汉族和少数民族群体的肠道微生物群比来自不同地点的同一少数民族群体的肠道微生物群更相似。基于功能预测,以小麦为主食的个体被预测拥有更多涉及葡聚糖1,3-β-葡萄糖苷酶和S-腺苷甲硫氨酸生物合成的微生物基因,而不是以大米为主食的个体。此外,观察到城市化对降低个体间多样性、增加个体间多样性和增加拟杆菌类型的比重有显著影响。总体而言,我们的研究提供了中国人群的全国性肠道微生物群基线和有关重要协变量的知识,这对于转化微生物群研究至关重要。

人类肠道拥有一个特殊的多样化微生物生态系统,据估计有150-400种细菌物种居住在我们的肠道中。肠道微生物群通过形成对抗病原体的屏障、产生生物活性代谢产物和调节免疫功能,为我们的健康提供了巨大的益处。肠道生态系统的稳态由一些核心物种维持,这些物种通常在不同个体之间共享,而在没有强烈影响因素(例如饮食变化或抗生素治疗)的情况下,健康成年人的肠道微生物群相对稳定。
肠道微生物群的失衡(即菌群失调)与许多疾病相关,例如炎症性肠病、肥胖症、过敏和自身免疫疾病。通过广泛的疾病靶向微生物群研究,已经揭示了许多微生物组分涉及一系列病理学,并且理论上可以作为生物标志物。例如,粪便微生物标志物用于筛选结直肠癌已经被广泛研究。然而,将微生物群研究转化为临床实践仍然受到多重挑战的限制,特别是精确分类“健康”微生物群的困难,这需要全面了解健康人群的微生物群变异和协变量。一项基于中国一个省份的14个地理区域的7009名个体的研究表明,在一个地方开发的基于微生物群的代谢疾病模型不能推广到其他地方,并且随着地理范围的增加,插值模型的效率降低。这强调了地理因素对肠道微生物群组成和疾病模型应用的影响,而中国更大地理范围内的微生物群变异尚未被探索。
除了地理因素,许多肠道微生物群协变量已经被揭示,包括饮食、生活方式、种族、社会经济地位、药物和遗传。例如,肠道微生物群的分层(称为肠道类型)已经与饮食,特别是纤维和碳水化合物的摄入有关;城市化已经与个体间变异的增加和具有高纤维降解潜力的物种的丧失有关;在种族间差异性丰富的类群被认为与慢性疾病有关。到目前为止,大多数研究肠道微生物群协变量的人口水平研究都集中在西方人群(欧洲和美国)以及以色列、日本和中国的少数研究,而在非洲、南美和亚洲其他地区的研究很少。
中国有各种食物风格,生活在不同地区的人在饮食上表现出很大的多样性。此外,中国有56个民族,这些民族在饮食、生活方式、习俗和文化方面具有独特的特点。因此,预计中国人群中的肠道微生物群会多样化。同时,由于城市化的速度和规模前所未有,中国人的生活方式正在迅速变化,饮食习惯正在向西式饮食转变。具体来说,消费更多的高脂肪和高蛋白食品,而摄入的谷物较少,这可能会显著改变肠道微生物群,就像在其他发展中国家观察到的那样。尽管已经进行了一些研究来调查中国肠道微生物群的特征,但这些研究要么关注有限的地区,要么招募了少数参与者,全国性的肠道微生物群调查仍然缺失。
为了表征中国人群的肠道微生物群多样性并研究与微生物群相关的变量,我们收集了2678名无明显疾病(称为“健康”)的个体的粪便,这些粪便经过了16S核糖体RNA(rRNA)基因测序(V3-4区域)。进行了包括人口统计学、饮食和生活方式信息的问卷调查,这使得可以深入分析与中国肠道微生物群相关的因素。

队列和数据概述
我们从28个省的63个县/市招募了2678名健康志愿者(男性1144人,女性973人),包括2167名汉族(1755名年龄超过3岁,412名年龄在3岁以下),487名来自七个少数民族(藏族156人;回族107人;苗族73人;维吾尔族70人;纳西族46人;蒙古族41人;白族18人),以及24名没有民族信息的个体(图1a,b)。粪便样本按照标准化程序收集(详见方法部分)。同时,通过问卷调查或国家年鉴收集了20个表型和环境变量,并将其归类为六个类别:地理、人口统计学、饮食、城市化、生活方式和采样月份(补充数据1)。通过测序16S rRNA基因的可变区域3-4(V3-4)来分析肠道微生物群,每个样本的中位数读数为27,638(范围10,000-236,350)。这些读数被聚类成14,364个零半径操作分类单元(ZOTUs),其中56.64%的ZOTUs(占总读数的86.48%)被分配到24个门下的444个属。
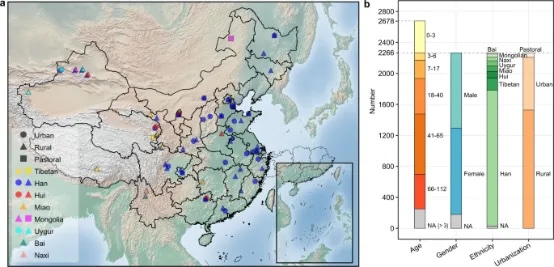
已知在3岁时会建立起一个类似于成年人的稳定肠道微生物群。在我们的数据中,我们观察到3岁以下儿童的α多样性与年龄之间存在强正相关(Shannon指数,R²=0.37;观察到的ZOTUs,R²=0.31;Faith的系统发育多样性(Faith's PD),R²=0.35,p<2.2e-16),但在其他年龄组中没有观察到这种相关性(3-17岁,18-65岁或66-112岁,p>0.05,补充图1)。因此,只有年龄在3-112岁之间的2266名个体(中位数46岁)被包括在随后的分析中。
中国人群肠道微生物群组成及相关协变量
厚壁菌门、拟杆菌门、变形菌门和放线菌门是所有样本中四个最丰富的细菌门(图2a,b)。共有24个属在>90%的样本中观察到,平均相对丰度>0.1%(核心微生物群,图2b)。这些属中有18个与中国广东省居住的2008名健康个体的核心肠道微生物群重叠(补充数据2);其中7个与另一个中国队列中前九个最丰富的粪便属重叠,该队列包括来自九个省的314名健康个体;其中10个与人类微生物组项目发现的前20个粪便属重叠。我们进一步使用Arumugam等人描述的聚类方法将微生物群细分为四种肠道类型。我们通过随机森林算法确定了驱动属(接收者操作特征(ROC)曲线下面积(AUC):0.99,补充图2a),得到了普雷沃特氏菌肠道类型(E1,n=443)、拟杆菌肠道类型(E2,n=732)、大肠杆菌肠道类型(E3,n=251)和混合肠道类型(E4,n=840)(图2c,d)。E1和E2是两个公认的肠道类型,而与大多数先前研究中作为第三个肠道类型的厚壁菌门(尤其是瘤胃菌属)不同,E3的特征是大肠杆菌(属于肠杆菌科,变形菌门)的过度表达,这很少被报道。此外,E4没有主导属,而是包括双歧杆菌和布劳特菌等一些相对丰富的属的混合。
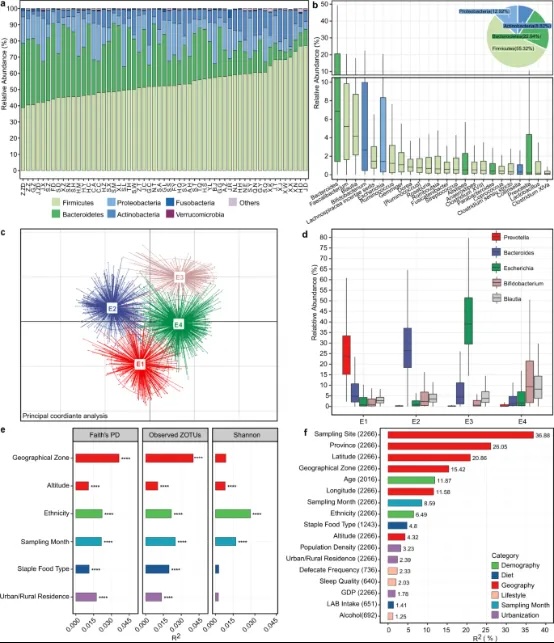
图2:中国人群肠道微生物群组成及相关协变量。a每个采样点前六个门的相对丰度。b 24个核心属的相对丰度,这些属在超过90%的个体中出现,平均相对丰度超过0.1%。饼图显示了队列的门水平微生物组成。LAB:乳酸菌。c 显示四种肠道类型的主坐标分析(PCoA)图。d 肠道类型代表性属的相对丰度。e 与微生物群α多样性相关的协变量。显示了经过调整的R²>0.01和****p<0.0001(简单线性回归)至少与一个α指数相关的协变量。f 通过Jensen-Shannon距离(JSD)估计的与微生物群β多样性相关的协变量。效应大小是通过envfit(vegan)计算的,显示了p.adj<0.05的协变量。每个协变量后的括号内标明了样本数量。在箱线图中,中心线代表中位数,箱线代表上四分位数和下四分位数,须线代表1.5倍四分位数范围。另见图S2。
首先通过简单线性回归调查了与肠道微生物群α多样性相关的协变量。Faith的PD(Faith的系统发育多样性)和观察到的ZOTUs(操作分类单元)与地理区域(包括十个不同气候、地形等的区域,补充图2b)、海拔、主食类型、城乡居住、民族和采样月份显著相关(调整后的R²>0.01,p<0.0001,图2e)。在包含所有六个协变量的多重线性模型中验证了这些相关性,除了与海拔的相关性(补充数据3)。这反映了海拔的依赖性,海拔在不同民族群体中是不同的(补充数据1)。此外,仅使用汉族个体的简单线性模型也不支持α多样性与海拔之间的相关性。
同时,肠道微生物群落结构(β多样性,通过Jensen-Shannon散度(JSD)估计)与17个协变量显著相关,这是通过envfit21评估的(p.adj<0.05,图2f)。地理因素(采样点、省份、纬度和地理区域)解释了最大的方差,其次是年龄、采样月份、民族、主食类型、城市化和其他地理因素(补充图2c、d为Bray-Curtis和未加权UniFrac距离)。为了进一步探索地理位置与肠道微生物群之间的相关性,我们在微生物JSD矩阵和地理距离矩阵上应用了Mantel检验,并发现它们之间存在显著相关性(p=0.03,补充图2e),这表明肠道微生物群在邻近地区逐渐变化。这些关联的详细分析在以下部分进行。
主食类型与肠道微生物群之间的关联
根据定期消费的主要主食类型,样本被分为三组,即大米(白米)、小麦(普通小麦的白面粉)和大米&小麦。由于不同谷物作物对温度、降水和日照时间的要求,小麦主要在中国北方种植,而大米的种植更为广泛。两种谷物的消费也显示出类似的地理分布(图3a)。有趣的是,包括Faith的PD、Shannon指数和观察到的ZOTUs在内的α多样性指数在消费更多大米的个体/地区显著更高(p<0.01,图3a,补充图3a-c)。
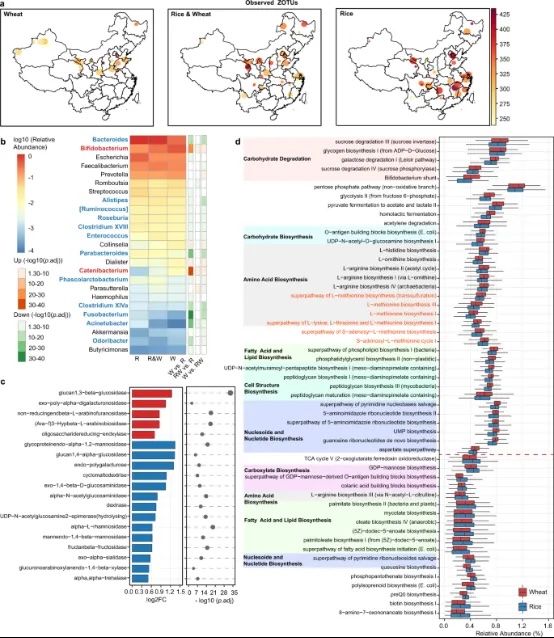
使用DESeq2分析并调整年龄和性别(p.adj<0.05,图3b),鉴定了两两组之间不同的细菌属。与食用大米的个体相比,食用小麦和小麦&大米的个体中,双歧杆菌属(Bifidobacterium)和链状细菌属(Catenibacterium)的丰度更高。这两个属的丰度在三组中逐渐减少,表明可能存在与小麦摄入量相关的剂量效应。双歧杆菌属与小麦摄入量之间的关联与先前的观察一致,即当采取低小麦含量的饮食时,例如无麸质饮食、低麸质饮食和低FODMAP(可发酵寡糖、二糖、单糖和多元醇)饮食时,双歧杆菌的丰度会减少23,24,25,26,27,28。与食用小麦的个体相比,食用大米和大米&小麦的个体中,有12个属的丰度更高,其中拟杆菌属(Bacteroides)、副拟杆菌属(Parabacteroides)、丁酸产生菌梭菌XIVa和机会性病原体梭杆菌(Fusobacterium)最为显著(log2FC>1,p.adj<1e-10)。然而,在三组中未观察到肠道类型的组成差异。
为了探索受不同主食影响的肠道微生物群的代谢能力,使用PICRUSt2对酶委员会(EC)编号和MetaCyc途径进行了推断。考虑到普通小麦粉含有的膳食纤维比白米多(干物质的2-3%对0.7-2%),并且两种谷物的纤维成分之间存在显著差异,我们特别关注属于糖苷酶(EC 3.2.1)的69个EC。小麦组和大米组之间有19个糖苷酶的丰度不同(log2FC>0.5,p.adj<0.05,图3c)。小麦组显示出显著更高的葡聚糖1,3-β-葡萄糖苷酶(EC 3.2.1.58),这与小麦而非大米中存在含β-(1->3)-键的β-葡聚糖的事实相一致。小麦组和大米组之间共有53条途径适度不同(log2FC>0.1,p.adj<1e-10,图3d)。首先,小麦组的特征是一些碳水化合物降解途径的潜力更高,以及糖酵解、磷酸戊糖途径和乳酸/醋酸发酵。其次,小麦组显示出包括L-甲硫氨酸、S-腺苷甲硫氨酸(SAM)和L-精氨酸等氨基酸生物合成能力的增加。值得注意的是,SAM广泛用作肝病、抑郁症和骨关节炎的治疗。此外,小麦组与包括细胞结构生物合成和核酸处理在内的管家功能潜力更高相关。这些发现表明,主食类型及可能相关的饮食习惯可能会改变肠道微生物群的代谢能力。
民族与肠道微生物群之间的关联
在本研究中包括的八个民族群体中,藏族的α多样性最高,而白族的α多样性最低(补充图4a)。此外,不同民族群体之间的肠道微生物群落结构不同(R²=4.00%,p<0.001,基于JSD的置换多变量方差分析(PERMANOVA),补充图4b)。然而,由于一些民族群体居住在特定的地理位置,很难将地理效应分离出来。在我们的研究中,四个少数民族群体,即维吾尔族、回族、蒙古族和藏族,不仅从不同地点收集了样本,而且还收集了来自同一地点的汉族样本,这使我们能够区分民族对微生物群的影响与地理的影响。对于所有四个少数民族群体,属于同一民族群体但来自不同地点(至少相距200公里)的样本之间的肠道微生物群丰富度(观察到的ZOTUs)存在差异。相反,来自同一地点的不同民族群体的微生物群丰富度没有显示出显著差异,除了维吾尔族和汉族之间(p.adj<0.05,图4a,补充图4c、d为Shannon指数和Faith的PD)。至于微生物群的β多样性,在主坐标分析(PCoA)图中,按采样地点和民族群体的聚类都可以区分(除了来自同一地点的藏族与汉族,p<0.05,基于JSD的PERMANOVA,图4b)。属于同一民族群体的样本的地点间距离大于来自同一地点的样本的民族间距离(维吾尔族、回族和蒙古族的p<0.0001),相应地,采样地点比民族群体在肠道微生物群中解释了更大的方差(PERMANOVA的R²:维吾尔族,7.70%对6.14%;回族,10.08%对3.81%;蒙古族,9.33%对4.63%;藏族,14.44%对4.88%)(图4b)。这些观察结果表明,地理和民族都可以影响肠道微生物群,但前者可能具有更强的影响。
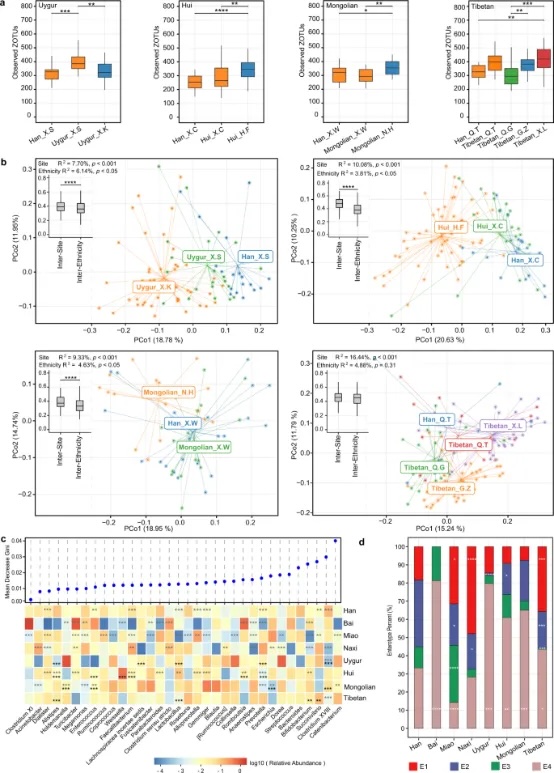
图4:不同民族群体的肠道微生物群特征。a 每个采样点不同民族群体的观察到的ZOTUs。x轴标签表示民族群体,后跟采样地点。*p.adj<0.05,**p.adj<0.01,***p.adj<0.001,Mann-Whitney检验。b 基于JSD的主坐标分析(PCoA)图。插入的箱线图显示了属于同一民族群体的样本的地点间距离,以及来自同一采样地点的样本的民族间距离;****p<0.0001,Mann-Whitney检验。也显示了来自PERMANOVA检验的相应R²和p值。c 不同民族群体之间优势属的丰度差异。属按随机森林模型分类民族群体时Gini平均下降值排序。显示了平均相对丰度>1%且在至少一个民族群体中超过50%的样本中存在的属。星号代表来自DEseq2模型的p.adj值;灰色的表示一个民族群体与其余群体之间的比较(并调整了采样地点),而黑色的表示每个少数民族群体(回族、蒙古族、藏族和维吾尔族)与同一采样地点的伴随汉族样本之间的比较;**p.adj<0.01,***p.adj<0.001。d 每个民族群体中肠道类型的百分比。*p<0.05,**p<0.01,***p<0.001,Fisher精确检验比较少数民族群体与汉族。在箱线图中,中心线代表中位数,箱线代表上四分位数和下四分位数,须线代表1.5倍四分位数范围。c-d 汉族:n=1755,白族:n=16,苗族:n=70,纳西族:n=46,维吾尔族:n=69,回族:n=87,蒙古族:n=40,藏族:n=154。另见图S4。
每个民族群体的属级微生物群特征显示出不同的相对丰度模式,我们能够使用随机森林模型区分不同的民族群体(模型的AUC为0.88;苗族0.94;维吾尔族0.93;白族0.93;藏族0.92;纳西族0.91;回族0.82;汉族0.80;蒙古族0.80;图4c,补充图4e)。链状细菌属(Catenibacterium)对分类的贡献最大,而肠道类型的代表性属,拟杆菌属(Bacteroides,E2)、大肠杆菌(Escherichia,E3)和普雷沃特氏菌(Prevotella,E1),分别排在贡献属的第5位、第8位和第9位。相应地,不同民族群体之间的肠道类型组成也不同(图4d)。与汉族相比,苗族、纳西族和藏族的E1比例更高;苗族、纳西族、维吾尔族、回族和藏族的E2比例更低;苗族的E3比例更高,而藏族的E3比例更低;除了纳西族以外的所有少数民族群体的E4比例更高(p<0.05,Fisher精确检验)。我们进一步应用DESeq2模型,通过将一个民族群体与其余群体进行比较来检测特定于民族的属,同时调整混杂因素采样地点;对于维吾尔族、回族、蒙古族和藏族,我们还比较了他们与同一采样地点的伴随汉族样本。两种模型都检测到的差异属(p.adj<0.05)包括维吾尔族中Clostridium XVIII的水平较低,拟杆菌属、费卡利布菌属(Fecalibacterium)和嗜粘蛋白艾克曼菌属(Alistipes)的水平较低,而回族中Romboutsia的水平较高,蒙古族中Holdemanella和肠球菌属(Enterococcus)的水平较高,而大肠杆菌的水平较低(图4c)。值得注意的是,一对其余的比较可能会受到不同民族群体之间不均匀的样本大小的影响。
城市化与肠道微生物群之间的关联
通过比较来自24个省的38个农村地点的1530名居民和来自18个省的22个城市地点的637名居民的肠道微生物群,我们发现农村居民的Faith的PD(系统发育多样性)高于城市居民,但观察到的ZOTUs(操作分类单元)和Shannon指数没有差异(图5a,补充图5a-c)。这表明城市化可能不会影响肠道微生物群的非系统发育丰富度和均匀度,但会减少其系统发育丰富度。同时,农村和城市居民的整体肠道微生物群组成不同(基于JSD的PERMANOVA,R²=1.63,p<0.01)。值得注意的是,通过JSD评估的城市居民的组内微生物群不相似性更高(p<0.001,图5b)。

图5:城市和农村人群之间微生物群多样性、组成和网络的差异。a Faith的PD(系统发育多样性)。每个点代表一个采样点;点的颜色表示每个地点的中位数;点的直径与每个地点的样本数量成比例,对于超过15个样本的地点是固定的。b 组间和组内JSD(Jensen-Shannon散度)。组内距离是按每个采样点计算的。****p<0.0001,Mann-Whitney检验。箱线图的中心线代表中位数,箱线代表上四分位数和下四分位数,须线代表1.5倍四分位数范围。c 使用DESeq2模型检测到差异属。显示了p.adj<0.05的属。d 属的共现网络。显示了r>0.35和p<0.01的SparCC相关性。每个节点代表一个属;节点的大小与相应属的中位数相对丰度成比例;绿色、红色和蓝色分别代表共有属、农村特有属和城市特有属。实线和虚线分别代表正相关和负相关,边缘的厚度与r值成比例。红色和蓝色星号表示c中显示的差异属,***p.adj<0.001。城市:n=637,农村:n=1530。另见图S5。
使用DESeq2分析比较了微生物群落,以确定区分城市和农村人群的属(p.adj<0.05,图5c)。在33个差异属中,最丰富的前两个(平均相对丰度>4.6%)是拟杆菌属(Bacteroides),它在城市人群中富集,而普雷沃特氏菌(Prevotella)则在农村人群中富集。此外,一些低丰度属(相对丰度<0.8%)在两组之间的差异更为显著(log2FC>1.5,p.adj<1e-20),包括在城市人群中丰度更高的Erysipelotrichaceae incertae sedis和Parasutterella,以及在农村人群中丰度更高的Alloprevotella和链状细菌属(Catenibacterium)。
除了上述微生物组成的差异,我们想知道微生物相互作用是否也因城市化而改变,因此分别为城市和农村人群构建了共现网络(图5d)。农村人群的网络比城市人群的网络更大(27个节点,36条边,与23个节点,23条边相比),并且将农村群体随机子采样到与城市群体相等的样本数量确认了这种差异。两个网络共享18个节点,但只有10条边共享,表明相同微生物对之间的相关性在两个人群之间是不同的。两个网络的枢纽节点也不同。在城市人群网络中,两个短链脂肪酸(SCFA)生产者,罗氏菌属(Roseburia)和费卡利布菌属(Faecalibacterium)(,以及与更多边连接的大肠杆菌(Escherichia),而在农村人群网络中,另外三个SCFA生产者,布劳特菌属(Blautia)、厌氧棒状菌属(Anaerostipes)和多尔菌属(Dorea),以及与更多边连接的Clostridium XVIII,表明两个人群的肠道生态系统中支持SCFA生产的不同生态组合。
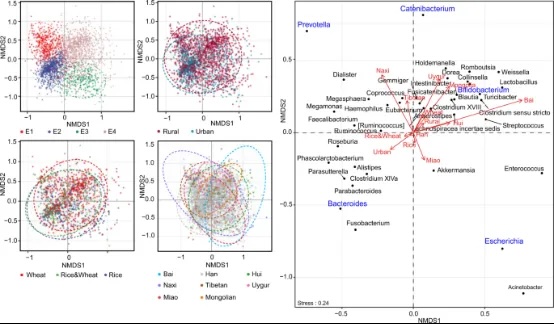
图6:非度量多维尺度图显示了按肠道类型、城乡居住、主食类型和民族群体分类的中国肠道微生物群的聚类。排序是基于前40个属的Bray-Curtis距离进行的。使用envfit(vegan)将协变量后期投影到图表上。椭圆代表95%的置信区间。

我们进行了一项迄今为止涵盖健康人群最大多样性的中国肠道微生物群研究。我们发现,属于五个类别的一系列因素,即地理、人口统计学、饮食、城市化和采样月份,解释了肠道微生物群变异的相当大部分,尽管某些因素(例如,民族)的效应大小可能因某些亚组中样本数量不均匀而被低估。首先,地理因素显示出最强的信号,包括采样地点、地理区域、海拔等。具体来说,来自同一采样地点的汉族和少数民族群体的肠道微生物群比来自不同采样地点的同一少数民族群体的肠道微生物群更相似,强调了在病例对照研究中考虑地理位置的重要性。尽管已有广泛报道肠道微生物群在地理上的差异,但由于地理反映了生活方式、长期饮食等混合效应,因此很难剖析这种效应。对于这个队列,我们未能阐明与地理区域相关的模式,而海拔效应很可能与居住在高原的少数民族群体有关。其次,我们关注了代表中国人口高度多样化的民族群体。汉族和七个少数民族群体显示出不同的肠道微生物群特征,其中一些变异归因于地理,而相当部分仍然可以显著地由民族群体解释。需要更大的队列和针对性的设计来理解民族群体背后的协变量(例如,遗传、习俗)对肠道微生物群的影响,以及这些效应在多大程度上被同汉族共同居住等因素同质化。此外,我们的发现强调城市化与肠道微生物群的个体内部多样性减少和个体间多样性增加有关。基于中国一个或两个省份的先前研究已经显示出细菌、真菌和病毒组分的类似模式15,37,38,而我们基于28个省份的研究进一步验证了城市化对中国各地肠道微生物群的巨大影响。此外,尽管采样月份似乎是一个显著的信号,我们没有发现α或β多样性中的任何特定季节模式。相比之下,狩猎采集者的肠道微生物群及其CAZYome多样性的季节节律已被显示。我们推测,由于现代人群中的生活方式,特别是饮食,受季节的影响要小得多,因此肠道微生物群与季节之间缺乏关联。
众所周知,饮食改变肠道微生物的组成和代谢,但对主食类型在人群水平上对肠道微生物组的长期影响的研究仍然缺失。中国人群主要消费两种不同类型主食,小麦(由普通小麦的白面粉制成的产品)和大米(煮熟的白米)。在这项研究中,消费小麦的人群被预测具有显著更高水平的编码葡聚糖1,3-β-葡萄糖苷酶的微生物基因,其底物仅存在于小麦而非大米中。这不仅证实了通过问卷获得的小麦摄入量的差异,而且证实了基于16S rRNA基因特征的功能预测的有效性。进一步地,小麦组中预测的微生物生物合成L-甲硫氨酸及其主要下游产物SAM的能力增加特别有趣,因为SAM在认知和代谢健康中的广泛参与。值得注意的是,在基于超高效液相色谱-质谱的尿液代谢组的干预研究中,已经显示了小鼠中微生物L-甲硫氨酸生物合成的改变以及SAM的古菌转化,该研究基于小麦的蛋白质成分,麦胶蛋白(gliadin),它可能在0.06%的中国人群中引发乳糜泻。因此,我们推测麦胶可能通过调节肠道微生物群,调节对麦胶敏感个体的健康,其有效性和机制需要进一步调查。此外,由于小麦在中国北方比南方更受欢迎,观察到的主食类型对肠道微生物群的影响可能被地理位置所混淆。
我们确定了上述因素显著影响的微生物群的特定组分。三个代表性的属,拟杆菌属、普雷沃特氏菌和大肠杆菌,推动了中国人群肠道微生物群的多样化(图6)。三个属的丰度在消费不同主食的亚群体中、在不同的民族群体中以及在城市与农村居民中有所不同。这可能是由于饮食习惯可能是这些因素背后的共同协变量,而拟杆菌属/普雷沃特氏菌与饮食习惯特别是纤维、蛋白质和动物脂肪之间的密切关系已被广泛显示。另一个值得注意的属是链状细菌属,它被发现在农村地区居住的人群中更丰富,以及在本研究中以小麦为主食的人群中。它也是这个队列中八个民族群体中最不同的属。链状细菌属在41%的人群中被检测到,平均相对丰度为0.6%。关于这个属的有限研究已将其与饮食联系起来,但结果相互矛盾。一些研究表明它与地中海饮食和低心血管疾病风险有关,而其他研究则显示它与高脂肪、高糖饮食有关。这些发现强调了需要进一步调查链状细菌属与饮食和人类健康的关系。此外,双歧杆菌与小麦摄入量有关,它也显著地促进了肠道类型的分化和八个民族群体的区分。值得注意的是,我们队列中双歧杆菌的总体丰度明显高于AGP队列和其他西方队列,这可能进一步加强了它在中国人群特定亚群体中的健康影响。这项研究的局限性在于缺乏全面生理指标和详细的饮食信息。这些信息对于理解这里揭示的单一肠道微生物群特征背后或受影响的因素至关重要,例如,在其他队列中很少报告的肠道类型大肠杆菌,在中国人群的特定子集中富集的链状细菌属,可能对人类健康产生影响的微生物SAM生物合成。尽管如此,获得的特征已经构建了中国肠道微生物群的基线,提供了微生物群变异、协变量和协变量效应大小的信息,这对于计算生物医学研究的样本大小和统计功率至关重要。此外,这项研究引起了在微生物群研究和临床转化中考虑微生物群背景和混杂因素差异的注意,包括通常适用的或特定于中国人群的因素。
本文译自:Lu J, Zhang L, Zhai Q, et al. Chinese gut microbiota and its associations with staple food type, ethnicity, and urbanization. NPJ Biofilms Microbiomes. 2021 Sep 6;7(1):71. doi: 10.1038/s41522-021-00245-0.
发表杂志:NPJ Biofilms Microbiomes.
通讯作者:Wei Chen
作者单位:Jiangnan University, Wuxi, China
