


人类肠道微生物组的多组学研究对于理解其在疾病中的多重功能层面的作用至关重要。然而,整合和分析这些复杂数据集带来了显著的挑战。尤其是,当前的分析方法通常会产生大量的与疾病相关的特征列表(例如,物种、途径或代谢物),但没有捕捉到数据的多层次结构。在这里,我们通过引入“MintTea”,一种基于中间整合的方法,结合典型相关分析扩展、共识分析和评估协议,来应对这一挑战。MintTea识别出“与疾病相关的多组学模块”,这些模块包括来自多个组学的特征,它们协调变化,并共同与疾病相关联。应用于不同的队列,MintTea捕获了具有高预测能力、显著的跨组学相关性和与已知微生物组-疾病关联一致的模块。例如,在分析一项代谢综合征研究的样本时,MintTea识别出一个包含血清谷氨酸和TCA循环相关代谢物的模块,以及与胰岛素抵抗相关的细菌物种。在另一个数据集中,MintTea识别出一个与晚期结直肠癌相关的模块,包括Peptostreptococcus和Gemella物种以及粪便氨基酸,这与这些物种的代谢活动和随着癌症发展的协调逐渐增加一致。这项工作展示了先进整合方法在生成微生物组-疾病相互作用背后的系统层面、多方面的假设方面的潜力。

人类肠道微生物组是一个极其复杂的生态系统,对我们的健康状况有着显著的多方面影响,并且在众多疾病的发病机制中扮演着已经确立的角色。对肠道微生物组的重要性和复杂性的认识促进了许多多组学微生物组研究的激增,这些研究对同一套样本应用了几种分子测定方法,希望能够捕捉到关于微生物组在疾病中作用的多层信息。在这些多组学研究中,一个越来越受欢迎的研究设计,例如,依赖于收集配对的高通量微生物组和代谢组分析(例如,结合霰弹枪宏基因组测序和质谱分析)。然而,不幸的是,尽管这样的多组学数据显然为研究微生物组及其在人类健康中的作用提供了一个激动人心的机会,但对这些数据进行严格的整合分析仍然非常具有挑战性,使用这些数据来获得对微生物组的系统层面理解也同样如此。
微生物组研究的一个共同目标,因此也是许多多组学微生物组分析的关键目标,是识别与疾病相关的标记物——来自不同组学的具体特征(例如,某些物种、途径或代谢物),其测量的丰度与所讨论的疾病有很强的关联。这样的标记物后来可以用来预测疾病,例如,指导基于微生物组的诊断,或者提出关于特定微生物组成分或机制的新假设,引导未来的实验和临床研究。鉴于疾病和对照样本(或者更一般地说,一些感兴趣的表型测量),可以使用各种统计方法来识别与疾病相关的标记物,这些方法从单独对每个组学中的每个特征应用的单变量统计测试,到考虑所有特征之间潜在统计依赖性的多变量统计方法,如偏最小二乘法(PLS)或线性回归。最近,机器学习方法也被广泛用于此目的,首先训练一些基于所有特征(无论是来自单一组学还是来自多个组合的组学)预测表型的机器学习模型,然后使用各种模型可解释性方法来检测模型中的信息特征。然而,最终,这样的分析通常会得到一长串与疾病相关的特征列表(来自一个或多个组学),没有利用多组学数据的多层次结构,也没有提供关于微生物组-疾病相互作用背后具体和连贯机制的清晰、可解释的假设。
已经引入了一些初步尝试来应对这一挑战,同时考虑跨组学依赖性和与疾病状态的关联。例如,一些微生物组研究采取了两步法,首先识别与疾病相关的特征,然后基于特征之间的成对相关性对这些特征进行聚类。然而,这种方法可能无法识别那些单独不够信息量的特征,但这些特征可以被整合到更大的模块中,作为一个整体,能够强烈预测疾病。其他旨在解决类似挑战的研究构建了跨组学相关性网络,同时在网络中包括了感兴趣的表型,或者为每种宿主状况构建了单独的网络,然后探索这些网络之间的差异以识别特定状况的模式。然而,由于组学数据的高维度,以及相应产生的庞大相关性网络,这些方法通常难以解释。
一种特别有趣的方法可能被用于解决这类分析,它源自多视角学习领域,被称为“中间整合”。与传统的多视角方法不同,传统方法直接结合原始特征,要么是通过天真地将不同的组学数据拼接成一个单一的联合表(即“早期整合”),要么是通过独立建模每个组学然后创建一个最终的集成模型(即“晚期整合”),中间整合试图将来自各种组学的特征(或“视角”)结合到一个中间表示层级,然后利用它们进行下游任务,如分类。这种方法因此捕捉了组学之间的依赖性,并且相应地有利于生成多方面的生物学假设。典型相关分析(CCA)例如,是一种流行的中间整合方法,它接收两个特征表,并输出每个表的线性转换,使得结果的潜在变量最大化相关。可能特别适用于微生物组多组学数据的CCA扩展包括稀疏典型相关分析(sCCA)——一种包含稀疏性约束以处理大量特征的CCA扩展,以及稀疏广义典型相关分析(sGCCA),它将sCCA推广以进一步支持超过两个视角。的确,CCA及其扩展以前已经应用于与微生物组相关的多组学数据,并取得了有洞察力的结果。例如,最近的一项研究将sCCA应用于患有肠易激综合征、炎症性肠病(IBD)或结直肠癌(CRC)患者的微生物组分类学特征和宿主转录组数据,识别了这些队列之间共享的宿主-微生物组关联以及其他特定于疾病的关联。在另一项研究中,sCCA被用来探索早期生活肠道微生物组和代谢组之间的关联,发现在6周龄时驱动这些关联的特定类群和代谢物与12个月龄时的不同。重要的是,CCA转换可以进一步考虑每个样本的疾病状态,从而寻找突出不同组学之间以及这些组学与疾病之间相互作用的表示。例如,Galié等人研究了地中海饮食对循环代谢物的影响,并使用sGCCA的扩展来识别饮食干预在肠道微生物组和血浆代谢物中共同反映的标志。这些研究证明了这种基于CCA的模型的潜在益处,然而,它们适用于不同的微生物组数据集,以及所获得结果的稳健性,仍然不清楚。此外,CCA及其扩展,以及类似的多元线性统计方法,通常对数据中的小扰动、参数选择(例如,稀疏性约束)、非典型样本和变量之间的共线性高度敏感,因此在应用于复杂的微生物组数据时需要仔细注意和解释。
为了解决上述挑战,并允许研究人员获得对微生物组-疾病相互作用背后连贯机制的系统层面的洞察,在这里,我们介绍了一种全面的基于中间整合的方法(结合CCA扩展、共识分析和验证协议),我们称之为“MintTea”,用于分析多组学微生物组数据。我们假设每种这样的机制可能涉及在疾病中协同作用的各种类群、功能和代谢物,因此MintTea旨在识别稳健的“与疾病相关的多组学模块”,每个模块都包含来自不同组学的特征集,这些特征不仅在不同组学间表现出协调的变化,而且作为一个整体与感兴趣的疾病或表型相关联。我们将MintTea应用于9个具有可用的霰弹枪宏基因组学数据的不同病例对照队列(处理成分类学和功能谱),其中6个还包括粪便或血清代谢组学数据。我们证明MintTea确实能够捕获具有高疾病预测能力的模块(通常与使用所有特征所达到的相当),同时表现出不同组学间特征的显著相关性。我们还展示了MintTea识别的一些模块重申了之前关于肠道微生物组在疾病中作用的观察结果,并提供了一个跨越所有分析数据集的已识别多组学模块目录。因此,这项工作作为先进整合方法在生成微生物组-疾病关联背后的整合多组学生物假设方面的益处的概念验证。

引入MintTea:一个用于识别稳健的与疾病相关的微生物组多组学模块的框架。
受到上述中间整合方法潜力的启发,我们开发了一个多组学整合框架,称为“MintTea”(微生物组分析的多组学集成工具),用于识别来自多个不同组学的一组特征,这些特征既与疾病强烈相关,也彼此之间强烈相关(图1A)。MintTea基于稀疏广义典型相关分析(sGCCA)以及之前介绍的其他相关方法,并且进一步应用重复抽样、共识分析和模块评估,以解决噪声数据问题并确保结果的稳健性和可信度。
简要来说,MintTea接收两个或更多的特征表,描述了同一套样本获得的不同组学,以及每个样本的标签(例如,健康与疾病),作为输入。在过滤掉稀有特征和其他预处理步骤之后(见方法),MintTea按照之前的建议,将标签编码为一个额外的组学(包含一个单一特征),然后应用sGCCA,寻找每个特征表的稀疏线性转换,以产生相应潜在变量之间的最大相关性,以及这些变量与标签之间的相关性(图1A顶部)。这个过程为每个特征表(组学)产生一个潜在变量,这是各种特征的稀疏线性组合。然后我们记录在各种组学中被赋予非零系数的特征集,并定义这个集合为第一个“假设模块”。sGCCA随后可以找到与先前潜在变量正交的额外潜在变量集,通过对那些先前潜在变量的缩减来实现,每次迭代同样提供一个新假设模块。接下来,为了识别对输入数据小变化稳健的特定特征模块,MintTea在随机数据子集(例如,样本的90%)上多次重复整个过程,并记录每次迭代产生的假设模块。然后它构建一个共现网络,如果特征在同一个假设模块中一致共现(例如,超过总迭代次数的80%),则特征之间相连,并识别出“共识模块”(即,连接的子图;见方法;图1A中部)。重要的是,由于MintTea在两个目标之间进行平衡,即组学之间的关联和与表型的关联,产生的共识模块可能只捕捉到这两个目标中的一个。因此,为了输出稳健的、与疾病相关的、跨组学相关的模块,MintTea最终评估每个共识模块,丢弃不符合期望标准的模块。为此,它首先过滤掉任何平均跨组学相关性不高于随机模块的模块。然后,MintTea对每个剩余模块中包含的所有特征应用主成分分析(PCA),使用每个模块的第一个主成分(PC)作为模块的代表,量化这个单一代表PC预测疾病的准确性,使用接收者操作特征曲线(AUC)下面积来衡量,并仅保留那些AUC > 0.7且高于随机模块(具有相同大小和组学分布;见方法;图1A底部)的模块。因此,MintTea的最终结果是多个“与疾病相关的、多组学模块”,每个模块捕获来自多个组学的特征,这些特征在不同组学之间高度相关,也与疾病相关。
在补充说明1中,我们讨论了MintTea与其他相关方法之间的概念差异,并对MintTea与其他基于sGCCA的方法进行了定量比较,证明MintTea在多个期望属性之间提供了良好的平衡,同时实现了显著降低的假阳性发现率。
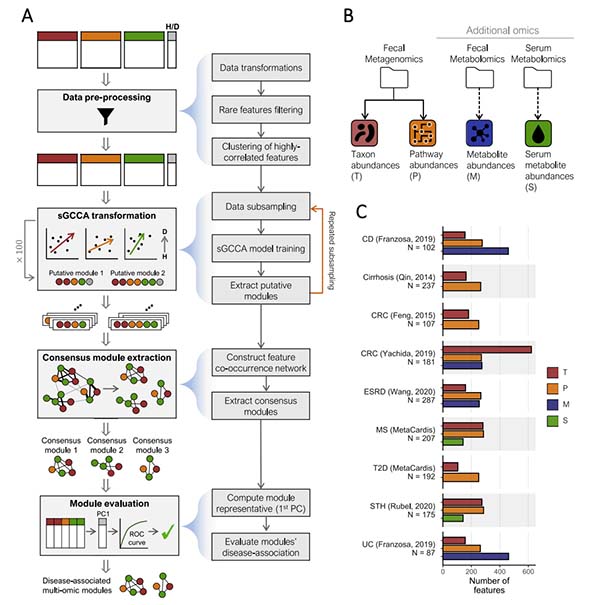
图1 | MintTea流程图示及本研究分析的多组学数据集。A MintTea流程的图示,包括数据预处理、使用稀疏广义典型相关分析(sGCCA)重复发现模块、共识分析,以及评估每个模块与疾病的关联。字母“H”和“D”分别代表“健康”和“疾病”。参见方法部分。B 在本分析中使用的数据类型。在整个手稿中,分类学、途径、粪便代谢物和血清代谢物特征分别用字母‘T’、‘P’、‘M’和‘S’标记。C 每个数据集在预处理后可用的特征数量。样本大小在数据集名称下方注明。CD克罗恩病,CRC结直肠癌,ESRD末期肾病,MS代谢综合征,T2D 2型糖尿病,UC溃疡性结肠炎。
使用MintTea在队列和疾病中识别多组学模块
为了检验MintTea在不同设置中识别与疾病相关的多组学模块的能力,我们获得了9个不同的霰弹枪宏基因组测序数据集。所有数据集都来自病例对照研究,涵盖了多种健康状况和疾病,包括克罗恩病(CD)、溃疡性结肠炎(UC)、结直肠癌(CRC)、代谢综合征(MS)、2型糖尿病(T2D)等。对于每个数据集,我们根据可用的组学数据生成了2-3个不同的特征表(“视图”)。具体来说,在所有数据集中,都有宏基因组数据,并处理成分类学档案(“T”)和途径级功能档案(“P”)(见方法)。此外,对于3个数据集,还有粪便代谢组学数据(“M”),而对于另外2个数据集,有空腹血清代谢组学数据(“S”)。图1B、C总结了用于主要分析的组学和数据集(另见补充数据文件S1)。

图2 | 通过MintTea中间整合框架识别与疾病相关的多组学模块。A-C 应用MintTea于几个关键数据集获得的多组学共识模块的特性,包括A 每个模块中的特征数量,按特征类型分层,B 每个模块的跨组学相关性,计算为不同组学特征之间的平均斯皮尔曼相关性(灰色点和线条表示从同等大小的随机模块获得的相关性的平均值和标准差),以及C 每个模块的AUC,使用每个样本的疾病标签对第一主成分值进行计算(灰色点和线条如前所述,表示从随机模块获得的相关性的平均值和标准差)。仅包含单一组学特征的模块已从我们的分析中排除,但在补充数据文件S2和S3中列出。展示在跨组学相关性高于随机模块(AUC > 0.7 且高于随机模块)的模块以较深的颜色显示。每个数据集名称下的圆圈颜色表示该数据集可用的组学。每个数据集的样本大小(即被分析的个体数量)如图1C所示。D 一个晚期CRC相关的多组学模块。节点颜色代表特征类型(参见图1B)。边连接在>80%的数据子采样迭代中一起出现在sGCCA潜在模块中的特征。模块内特征对之间的相关性可以在补充数据文件S4中查看。E 一个与MS相关的多组学模块。
将MintTea应用于这9个数据集,我们发现了每个数据集的2-5个共识模块,每个模块包含来自不同组学的2到38个特征(平均:13.23;图2A;补充数据文件S2;补充数据文件S3)。这些模块中的大多数捕获了不同组学之间特征的相关性,这些相关性显著超过了随机抽样模块的相关性(图2B),每个数据集有1-2个模块也表现出与疾病强烈的关联,如上定义(图2C;补充数据文件S2和S4)。
例如,在T2D(MetaCardis13,53)数据集中,我们确定了5个共识模块,其中2个也通过了我们的评估,并被归类为与疾病相关的多组学模块,AUC值达到0.86(基于该模块中21个特征的第1个主成分)和0.73(基于该模块中仅有的2个特征),与随机选择的模块相比,平均分别为0.71和0.64。在晚期CRC数据集(Yachida, 201911)中,作为另一个例子,我们发现了2个共识模块,其中一个被归类为与疾病相关的多组学模块,AUC为0.72,相比之下,使用等效的随机选择模块仅为0.63(关于这个模块下面有更多信息)。所有已识别模块的完整列表,以及它们与疾病关联和跨组学特征相关性的详细统计数据,都提供在补充数据文件S2-S4中。
虽然我们下面的大多数分析只关注与疾病相关的多组学模块(即,通过了我们的评估阶段并符合我们定义的标准的模块),我们也试图评估每个数据集中所有已识别共识模块的整体预测能力,以更好地了解基于CCA的方法识别数据中信息变异轴的能力。为此,我们在每个数据集中所有模块的第1个主成分上训练了随机森林(RF)模型,并估计了它们的组合预测准确性(带交叉验证)。我们进一步将这些模型的AUC与在完整连接的多组学数据上训练的RF模型的AUC进行了比较(称为“早期整合”方法;见方法;补充数据文件S5)。我们发现在大多数情况下,这些共识模块本身实现了与早期整合模型相当的AUC,同时使用的总特征数量要少得多(补充图S2)。例如,在UC(Franzosa, 201912)数据集中,MintTea识别的4个共识模块(总共73个特征)达到了0.885的总AUC(标准差:0.12),其中每个模块由其第1个主成分表示,如上所述。相比之下,早期整合方法达到了类似的AUC 0.882(标准差:0.14),但在特征选择后平均使用了235个特征来训练RF模型。同样,CRC(Feng, 2015)54模块的整体AUC为0.839(标准差:0.15),仅在3个模块中使用了36个特征(因此基于仅有的3个值),相比之下,基于早期整合的103个特征的AUC为0.824(标准差:0.15)。
值得注意的是,上述报告中与疾病相关的多组学模块所包含的特征,并不一定与早期整合的多组学模型中排名最高的特征重叠。具体来说,平均而言,每个与疾病相关的模块(如上所定义)包含的特征中,只有63.9%(标准差:24.8%)也是多组学早期整合模型的重要贡献者(参见方法,补充数据文件S6)。这一观察可能证明了中间整合方法的能力,即考虑特征在与其他特征的关联背景下的作用,而不仅仅是它们与疾病的关联。
与疾病相关的多组学模块揭示了多方面的生物学特征
图2D展示了在晚期CRC数据集中识别出的MintTea模块的一个例子。这个模块总共包括了7个特征;3种细菌物种,即厌氧消化链球菌(Peptostreptococcus stomatis)、无氧消化链球菌(Peptostreptococcus anaerobius)和摩比尔拉氏菌(Gemella morbillorum),以及4种粪便代谢物:2种支链氨基酸(BCAA's),即缬氨酸(Val)和亮氨酸(Leu),一种芳香族氨基酸 - 苯丙氨酸(Phe),以及半胱氨酸-谷胱甘肽二硫化物二价体。这个模块中包含的7个特征的第1主成分(PC),单独使用,就在分类疾病状态时产生了0.72的AUC(图2C),不同组学特征之间的平均斯皮尔曼相关性为0.28(所有相关性的FDR校正p值均小于0.05,补充数据文件S4)。除了最后一个特征外,这个模块中的所有特征在疾病状态下都显著升高(Mann–Whitney测试,FDR < 0.05),并且在多项其他独立研究中也被报告为疾病生物标志物。特别是,在Yachida等人的原始论文中,这三种物种在所有疾病阶段中一直过度表示,与其他只在特定阶段升高的物种相反。三种氨基酸(Val、Leu和Phe)也报告了类似的趋势,表明与上述分类群存在协调的模式。在那项研究中,厌氧消化链球菌和摩比尔拉氏菌也被发现在所有癌症阶段与对照组相比具有显著高的复制率,暗示它们在癌症发展期间的代谢活性增加。厌氧消化链球菌物种与BCAA's一起出现的一个可能解释是它们参与了BCAA的代谢。有趣的是,厌氧消化链球菌物种被特别识别为Phe和Leu的主要发酵菌,而最突出的CRC相关物种,梭杆菌(Fusobacterium nucleatum),相比之下,在同一篇综述研究中被报告偏好不同的氨基酸底物。苯丙氨酸在最近的一项元分析中也与厌氧消化链球菌和摩比尔拉氏菌显著正相关。最后,尽管我们无法恢复半胱氨酸-谷胱甘肽二硫化物二价体与其他特征之间的机制联系,但我们注意到,即使在控制疾病状态的情况下,这种代谢物与另外3种氨基酸之间的相关性也非常高(所有斯皮尔曼和偏斯皮尔曼相关性ρ > 0:47,FDR < 1 X10^-10)。
另一个例子,如图2E所示,是在MetaCardis的代谢综合征(MS)队列中识别出的模块(这些是根据国际糖尿病联盟标准被诊断为MS的对象,没有2型糖尿病(T2D)或冠状动脉疾病)。这个模块包括8个物种(其中2个缺乏分类学注释)、4个途径和9种血清代谢物(通过核磁共振或质谱测量),这些特征的第1主成分(PC)达到了0.72的AUC,用于预测MS。值得注意的是,与粪便代谢物相比,肠道微生物组与血清代谢物之间的相互作用显著不那么直接,因为它可能被其他生理过程和系统代谢活动所掩盖。实际上,这个模型中不同组学特征之间的平均相关性低于其他数据集模块中的相关性,但仍然高于随机抽样的模块(图2B)。具体来说,在这个模块中,108个血清代谢物与肠道微生物组特征之间的相关性有19个是显著正相关的(FDR < 0.05;相比之下,在同样大小的随机模块中,不到1个代谢物-微生物组特征对是显著相关的)。在这个模块中包含的血清代谢物中,有几个是众所周知的代谢紊乱的标志,包括升高的葡萄糖、甘露糖、三羧酸循环代谢物(例如丙酮酸和α-酮戊二酸)以及谷氨酸。循环中的异亮氨酸水平(以及其他支链氨基酸和相关代谢物)也反复被证明在葡萄糖稳态和代谢综合征风险中发挥作用。尽管这个模块中的一些其他代谢物在代谢紊乱的背景下没有被特别讨论,但它们彼此之间都显著正相关(36对代谢物中有32对正相关,FDR < 0.05),有效地代表了在MS中协调升高的一组代谢物。有趣的是,在最近的一项研究中,发现这些代谢物中的几种(丙酮酸、葡萄糖、谷氨酸)在粪便中测量时,与肠道微生物组显著相关,但在血液中测量时预测效果只是勉强好,这进一步加强了肠道微生物组与这些血清代谢物的关联可能被其他过程所掩盖,但仍然存在。
此外,这个模块中某些细菌物种与这些代谢物的结合可能与它们先前报道的在谷氨酸代谢和/或支链氨基酸(BCAA)代谢中的作用有关。例如,虽然在我们的数据集中,拟杆菌属(Bacteroides)的dorei/vulgatus在MS患者中只是边际显著增加(FDR = 0.07),但已被证明会影响血清异亮氨酸水平。特别是,无氧消化链球菌(B. vulgatus)也被报道参与谷氨酸代谢,并进一步被认为是驱动肠道BCAAs生物合成与胰岛素抵抗之间关联的主要物种之一。然而,无氧消化链球菌在代谢紊乱发展中的确切作用仍然不清楚,可能依赖于特定的环境和菌株,因为它在一些研究中被证明能带来健康益处(包括与代谢相关的),而在其他研究中则与胰岛素抵抗和体脂呈正相关。有趣的是,无氧消化链球菌还被报道在给予大鼠时会引起组成变化,特别是促进了这个模块中也包含的拟杆菌属(Parabacteroides)物种的增加。这个模块中的另一个物种,拟杆菌属(Parabacteroides mardeae),也被证明能增强BCAA的分解代谢,并再次带来积极的健康益处,尽管在我们的数据集中在MS患者中显著增加。最后,我们注意到,沃氏拟杆菌(Bilophila wadsworthia)在文献中大多在IBD和结肠炎症的背景下被讨论,但也被牵涉到代谢功能障碍中。它还被发现在对短期和长期饮食的反应上与另一模块中的物种,梭菌属(Clostridium bolteae)聚集在一起,可能解释了它们在这个模块中的共同出现。至于这个模块中包含的途径特征,可能并非所有都代表了与MS相关的特定代谢功能,但仍然与模块中的一些物种表现出强烈的相关性,表明了潜在的依赖关系。总的来说,我们发现有多个报告支持模块内的特定联系,但这些细菌物种在MS表型和MS相关代谢物的循环水平中的共同作用仍然难以捉摸。
重要的是,即使仅分析来自宏基因组的特征(即在没有额外组学数据的情况下),MintTea也可能突出与相关的分类学和功能性疾病生物标志物有关的有趣见解。例如,在肝硬化数据集中,我们发现了一个模块,该模块包括了通常在人类口腔中发现的多种物种,包括几种链球菌(Streptococcus)、韦永氏菌(Veillonella)和巨球形菌(Megasphaera)物种(包括共生菌和机会性病原体),所有这些物种都先前被发现在患有肝病的个体的粪便样本中增加,以及其他胃肠疾病。这个模块中多种链球菌和韦永氏菌物种的共同出现与它们强烈的共同丰度模式一致,这一点在多个不同的大型队列中得到了确认,并且也与它们之间已知的代谢相互作用一致。此外,模块中的几个途径与维生素K代谢有关(如甲萘醌-8生物合成I、甲萘醌-11生物合成、2-羧基-1,4-萘醌生物合成、叶绿醌生物合成、生育三烯二磷酸生物合成),这些也与肝脏健康相关。实际上,已知几种维生素K形式(包括甲萘醌)是由人体肠道中的细菌产生的,特别是韦永氏菌被报告在体外产生甲萘醌。在另一项研究中,发现甲萘醌生物合成与IBD中链球菌过度生长有关。模块中的其他途径,例如与L-甲硫氨酸和血红素b生物合成有关的途径,也可能与甲萘醌代谢和韦永氏菌特定的代谢活动有关。例如,L-甲硫氨酸作为某些甲萘醌生物合成的甲基供体的作用已在几种细菌物种中得到确认,尽管不是在这个模块的物种中。此外,对韦永氏菌物种的先前基因组分析表明,它们拥有完整的血红素生物合成基因集。总的来说,这个模块表明了一个潜在的多层次的与维生素K相关的、由口腔细菌驱动的机制,与肝损伤高度相关。补充图S3进一步展示了与IBD相关的两个额外的多组学模块,作为额外的例子。
多组学模块在不同队列中的共享
在展示了MintTea能够识别展现出跨组学关联以及与疾病相关的多组学模块之后,我们最终寻求检测在不同数据集中共享的普遍和反复出现的多组学模块。由于分析的多组学数据集之间存在显著的技术和方法差异,这使得跨研究比较变得不切实际,因此我们在这里仅关注霰弹枪数据,其中分类学和功能(途径级别)档案是由curatedMetagneomicData资源统一生成的。具体来说,我们使用了之前介绍的来自curatedMetagnomicData资源的3个数据集(即CRC、肝硬化和STH数据集;图1C),以及来自同一资源的11个额外数据集(补充数据文件S1;方法),共计14个统一处理的数据集。在对这些数据集应用MintTea之后(补充图S4;补充数据文件S3和S4),我们量化了来自不同数据集和疾病状态的模块之间的重叠,并识别出显著的重叠(图3A;补充数据文件S7;方法)。在至少有2个特征重叠的43对共识模块对中,有10对展现了统计学上显著的重叠(费希尔精确检验,FDR校正p值<0.1),这表明微生物组特征之间存在普遍的多组学关联。
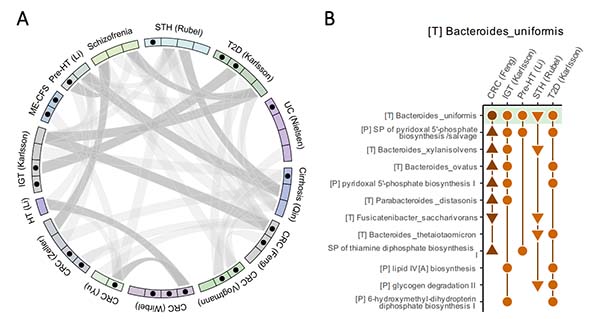
图3 | 跨数据集的多组学模块。A 展示了在至少一个其他模块中出现的多组学特征之间的重叠。三角形指向不同数据集的模块。马戏团图中的每个扇区代表在该模块中疾病状态下特征水平显著增加的特征,按它们所属的数据集进行分组。黑色点代表数据集,而指向下的三角形表示相反的趋势(Mann–Whitney测试,FDR < 0.1)。圆圈表示研究组之间没有显著差异。连接不同模块的线条表示至少2个特征的重叠,较深的线条表示统计学上显著的依赖关系(费希尔精确检验,FDR < 0.1)。B 包括B. uniformis的多个数据集的多组学模块,以及额外的重叠特征。对于每个模块(顶部列出的数据集),所有模块中的物种和途径都以不同的颜色编码。有关缩写,请参见图1的图例。
我们还发现了特定的细菌物种出现在不同数据集的多个模块中。例如,拟杆菌属(Bacteroides)uniformis——一种在人类胃肠道中高度丰富、能够降解纤维的革兰氏阴性共生菌——出现在5个不同数据集的模块中。与B. uniformis一起出现在MintTea模块中的其他常见物种也来自拟杆菌属,包括B. xylanivsolvens、B. ovatus和B. thetaiotaomicron。有趣的是,B. uniformis倾向于与维生素B相关的途径一起出现,如维生素B6的活性形式——吡哆醛5'-磷酸生物合成途径和硫胺素(维生素B1)二磷酸合成途径。实际上,已知B. uniformis基因组以及其他拟杆菌属基因组具有多种维生素的生物合成途径的相对高覆盖率,包括B族维生素。此外,在之前的一项研究中,我们发现拟杆菌属与粪便中的吡哆胺水平(一种维生素B6的形式)显著且一致地相关,这是基于对5个不同数据集的元分析,其中既检测到这个属也检测到这种代谢物。虽然这项分析仅在属级别进行,我们也知道B. uniformis、B. thetaiotaomicron和B.ovatus(如图3C所示)是成人中最普遍的拟杆菌属物种之一。最后,通过检查HUMAnN3的分类学分层输出,我们观察到B. uniformis是分配给这些维生素B相关途径的读取量的主要贡献者之一,进一步解释了它们在同一模块中的共同出现。B. uniformis也是其他与它一起出现在来自不同数据集的模块中的途径的主要贡献者,包括6-羟甲基二氢蝶呤二磷酸生物合成I和脂质IVA生物合成(这是脂多糖(LPS)生物合成过程的一部分,LPS是细菌外膜的关键组成部分)。综合来看,这些模块表明了一个在多个队列之间共享的共同变异轴,主要由拟杆菌属物种和对细胞存活至关重要的代谢过程所特征,包括几种辅因子和LPS的生物合成。
注意到上述拟杆菌属物种倾向于在同一模块中共现的趋势,我们进行了基于排列的分析,以测试MintTea模块中的物种是否确实比随机模块中的物种更有可能与其各自属中的其他物种共同出现。我们确认这确实是一个普遍趋势(22.8%与12.7% [7.7–18.6%]相比,p值:0.001)。这一观察结果与以下假设一致:系统发育相关的物种更有可能为共享的生态位竞争,因此表现出更强的共现模式。

在这项工作中,我们着手增强微生物组多组学数据的分析,使研究人员能够识别跨越多个分子过程的稳健的、与疾病相关的微生物组特征,并生成关于微生物组在疾病中作用的多方面假设。我们特别采用了一种“中间整合”方法来分析多视角(多组学)的人类肠道微生物组数据,提出了一个框架(称为“MintTea”),用于同时识别来自不同组学的高度相关联的特征集,这些特征集不仅彼此高度相关,而且与研究中的疾病共同相关。我们的“视角”包括从射枪宏基因组测序数据中得出的分类学和通路功能剖面,以及代谢组剖面(来自粪便或血清)。应用于多个不同的病例对照数据集,MintTea能够精确识别出反映跨组学数据协调变化的连贯多组学模块,同时也高度预测疾病。
使用MintTea,例如,我们识别了一个由Peptostreptococcus和Gemella物种以及特定氨基酸的代谢驱动的CRC(结直肠癌)相关模块。在另一个代谢综合征患者的队列中,我们发现了一个模块,其中包括一组高度相关的空腹血清代谢物(主要是谷氨酸和TCA循环相关的代谢物)以及之前与胰岛素抵抗/代谢紊乱以及异亮氨酸代谢有关的细菌物种。将MintTea应用于大量射枪宏基因组数据集,并通过比较每个数据集中识别出的共识模块,我们进一步识别出多个具有显著高重叠的模块,这表明肠道微生物组的组成和功能在多个队列中共有常见的变异轴。未来的工作可以特别搜索不同队列中相同疾病的共享模块,或者跨越额外组学(或临床数据,例如,来自电子病历)的共享模块,以获得更多与临床相关的关于疾病相关微生物组过程的见解。由于数据可用性的限制,这个提议的分析仍然不在当前工作的范围之内。
总的来说,中间整合方法可以作为早期整合方法的一种替代(或补充)分析方法,早期整合方法在应用某些机器学习模型之前只是简单地将不同的组学数据串联在一起(我们在这项研究中使用了这种方法作为疾病可预测性的基线)。实际上,随着多组学研究设计的增多,中间整合方法近年来越来越受到关注,因为它们能够揭示复杂的多变量相互作用,这在处理复杂的生物系统时是非常必要的。我们使用基于sCCA的方法的动机是假设肠道微生物组与疾病之间的关联可能在微生物组多组学数据中以一个或多个“模块”的形式表现出来,这些模块中的所有组学特征都参与了由其效应或促进宿主疾病的特定机制。此外,基于sCCA的框架以前已经应用于各种微生物组多组学数据,并取得了有洞察力的结果。然而,为了克服数据高维度和结果不稳定性带来的挑战,我们通过使用数据子样本的重复迭代和共识分析进一步扩展了这种分析技术,以基于多个基于sCCA的假定模块识别出稳健的共识模块。这种保守的方法确实大大降低了假发现的风险,同时在与其他评估指标相比时保持了类似的性能,当与类似方法比较时(见补充说明1)。
本质上,MintTea是一个数据驱动的假设生成工具,旨在发现由多层信息反映的微生物组特征。然而,这些特征的评估并不简单,它是一个开放的挑战,也是多重整合方法的一个主要限制。我们特别认识到三种有价值的评估类型:首先,稳健性评估,即在数据扰动或方法参数变化的情况下,结果的一致性。MintTea通过要求模块在多次数据扰动(例如,抽样)中保持稳定,从而内在地提高了稳健性,实际上,这种增加如前所述,产生了较低的假发现率。第二种评估可能涉及所获得发现的普适性,即在独立队列中复制结果的能力。我们研究的一个限制是缺乏这种跨研究验证多组学模块,这是由于从不同队列获取统一的多组学数据所涉及的挑战。我们注意到,随着多组学数据共享变得更加普遍和标准化,这种验证将变得至关重要。最后,也许最重要的是,结果应该理想地评估其生物学的连贯性和相关性。实际上,这代表了MintTea(以及其他纯粹基于统计的多组学整合方法)的一个主要警告。不幸的是,系统地验证推断出的多组学特征之间的相互作用的意义是一项挑战,这在很大程度上是因为缺乏或未知的这种多组学机制的系统映射。因此,我们鼓励MintTea的用户谨慎对待结果的解释。
我们还注意到,任何基于宏基因组和其他特征来识别与疾病相关的模块的尝试,其准确性和可靠性只能取决于这些底层特征本身。尽管有几种已经建立起来的宏基因组数据分类学和功能分析工具,但它们每个都可能引入偏差和不准确性。此外,可能还可以从宏基因组学和代谢组学数据中恢复额外的、可能是非常宝贵的特征,例如,通过使用基于组装的方法来增加未知基因组的丰度或将菌株水平信息添加到分类学分析中,或者从非靶向代谢组学分析中添加未注释的代谢物。另一个限制是对已知会引起微生物组变化的主要混杂因素(例如,粪便一致性、饮食和药物使用)的控制不足。通过仔细的样本分层、将这些因素作为协变量包含在模型中,或者甚至将这些数据作为额外的“视角”添加进来,可能会潜在地缓解这一限制。例如,饮食很可能是许多已识别的微生物组-代谢组模块的重要中介,因此将“饮食”-视角剖面包含在未来的分析中以阐明潜在的饮食驱动模块,可能会特别有趣。最后,由于微生物组组学数据具有独特的特性,线性方法如CCA可能不足够,应探索和发展替代的算法和统计方法。
总体而言,这项工作证明了应用先进的中间整合方法到微生物组多组学数据中,用于生成关于微生物组在人类疾病中作用的多方面假设的潜在益处。我们提倡使用这种多组学中间整合模型来捕捉微生物组-疾病关联的互补生物学见解。
本文译自:Muller E, Shiryan I, Borenstein E. Multi-omic integration of microbiome data for identifying disease-associated modules. Nat Commun. 2024 Mar 23;15(1):2621. doi: 10.1038/ s41467-024-46888-3.
发表杂志:Nat Commun
影响因子:16.6
通讯作者:Elhanan Borenstein
作者单位:Tel Aviv University, Israel
