

背景:
越来越多的证据表明,肠脑轴的紊乱可能是导致重性抑郁障碍(MDD)的潜在原因之一。然而,抗抑郁药对肠道微生物的影响,以及肠道微生物在影响抗抑郁药效果中的作用,仍然没有得到充分的理解。
结果:
为了填补这一知识空白,我们进行了一项多组学研究,涉及110例接受舍曲林(ESC)治疗12周的MDD患者。这项研究在一个队列中进行,并与166例健康个体的参照组进行了比较。结果发现,ESC通过上调MDD耗竭的氨基酸和下调MDD富集的脂肪酸,改善了异常的血液代谢。另一方面,ESC对肠道微生物的抑制作用相对较弱,导致微生物丰富度和功能的降低。基于机器学习的多组学整合分析揭示了肠道微生物对血浆代谢物的变化有贡献,并与若干氨基酸,如色氨酸及其肠道微生物衍生的代谢物吲哚-3-丙酸(I3PA)有关。值得注意的是,在第12周的基线微生物丰富度和临床缓解之间观察到了显著的相关性。与未缓解者相比,缓解者的基线微生物丰富度更高,菌群失调评分更低,而且他们的微生物群落结构和细菌网络更复杂和有序。这些发现表明,缓解者的微生物群落更具有恢复力。此外,我们还证明,预测ESC治疗后临床缓解的可能性的不是肠道微生物的组成本身,而是基线时存在的芽孢形成基因。基于这些基因的预测模型显示了一个曲线下面积(AUC)性能指标为0.71。
结论:
这项研究为肠道微生物在ESC治疗MDD患者的效果机制中的作用提供了宝贵的见解。这些发现代表了在理解抗抑郁药、肠道微生物和血液代谢组之间的复杂关系方面的重大进展。此外,这项研究提供了一个以微生物为中心的视角,可以潜在地提高抗抑郁药在临床实践中的效果。通过揭示这些因素之间的相互作用,这项研究有助于我们对抑郁症的更广泛的理解。
引言:
我们对110例MDD患者的粪便和血浆样本进行了纵向的多组学分析,以探讨舍曲林(ESC,一种SSRI抗抑郁药)对肠道微生物的影响,以及肠道微生物与ESC效果的关系。首先,我们对抗抑郁药治疗12周期间肠道微生物组成、微生物功能以及粪便和血浆代谢物谱的纵向变化进行了全面的分析。接下来,我们研究了肠道微生物在介导抗抑郁药缓解中的作用和贡献。最后,我们探讨了基线肠道微生物作为临床抗抑郁药缓解的预测因子的潜力。通过分析治疗前个体的初始微生物组成,我们试图确定是否有特定的微生物标志物或模式可以作为达到抗抑郁药治疗缓解的可能性的指示器。
结果:
1、实验设计
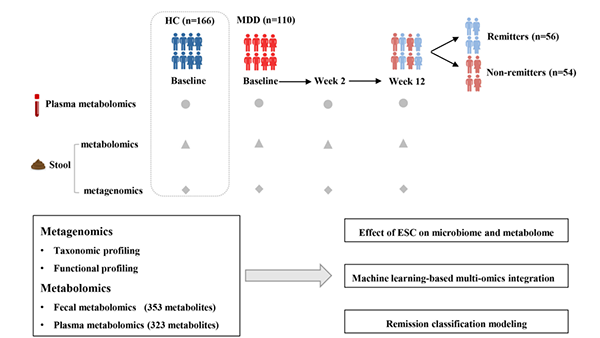
图1 研究设计概述。
本研究的工作流程示意图展示了样本组和多组学数据收集的细节。MDD患者接受ESC治疗12周,治疗结果在第2周(早期)和第12周(终点)进行评估。所有患者根据第12周的HAMD-17评分分为缓解者(R,HAMD-17≤7)和未缓解者(NR,HAMD-17>7)两组。
1、ESC治疗改善了MDD患者的血浆代谢物失调
MDD患者的总体血浆代谢特征与HC组有显著差异,如主成分分析(PCA)所示(PERMANOVA,Euclidean距离,p=0.001,图2A)。与HC组相比,MDD组显示出7种代谢物的富集和37种代谢物的耗竭(q<0.1;图2B)。MDD患者的特征是有机酸代谢物(其中大部分属于脂肪酸)的水平较高,如3-羟基丁酸、棕榈油酸、2-羟基丁酸、棕榈酸和亚油酸,以及氨基酸代谢物的水平较低,包括L-酪氨酸、L-苯丙氨酸、L-丙氨酸、L-缬氨酸、L-脯氨酸、L-蛋氨酸和谷氨酸。
ESC治疗后,如图2C所示,多种氨基酸代谢物被上调,包括L-酪氨酸、L-蛋氨酸、L-苯丙氨酸、L-丙氨酸和L-色氨酸。一个值得强调的重要观察是,L-色氨酸是神经递质5-羟色胺(5-HT)的前体,通常被称为血清素。L-色氨酸具有通过血脑屏障的能力,色氨酸的血浆水平通常被认为是中枢5-HT活性的可靠指标。有趣的是,尽管进行了ESC治疗,我们并没有观察到血清5-HT水平的增加。然而,我们的发现显示,另一种来自肠道微生物的色氨酸代谢物,吲哚-3-丙酸(I3PA),被显著地上调。I3PA最近被报道能够促进神经再生和修复。相反,有机酸(其中大部分属于脂肪酸)被显著地下调,包括硬脂酸、油酸、亚油酸、2-羟基丁酸、棕榈油酸、4-脱氧赤藓糖酸、3-羟基丁酸、苯酚和棕榈酸。Fisher精确检验显示,上调的代谢物富集在HC富集的代谢物集合中,而下调的代谢物富集在MDD富集的代谢物集合中(p=6.73e-05)。值得注意的是,6种上调和2种下调的代谢物与HC富集的代谢物重叠,而0种上调和6种下调的代谢物与MDD富集的代谢物重叠。综上所述,这些发现暗示,ESC治疗通过上调氨基酸代谢和下调脂肪酸代谢,改善了MDD患者的血浆代谢物失调。
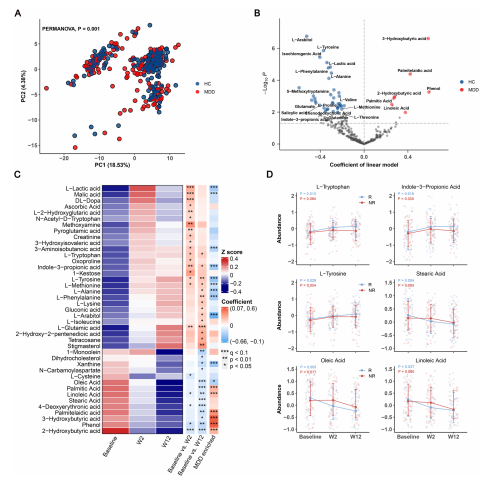
图 2 MDD患者的血浆代谢组失调及ESC治疗对血浆代谢物的影响。
a)基于血浆代谢组谱的主成分分析(PCA)显示,MDD患者和HC组的血浆代谢特征之间存在显著差异(PERMANOVA,欧氏距离,P=0.001)。
b)MDD患者和HC组之间代谢物水平差异的回归系数的火山图。多元回归模型用于探索血浆代谢物与疾病状态之间的关联,统计显著性由q值<0.1确定。与HC组相比,MDD组的特征是7种上调的代谢物和37种下调的代谢物,这些代谢物主要涉及有机酸和氨基酸。
c)显示了41种血浆代谢物的平均丰度的热图,这些代谢物在ESC干预后的第2周或第12周发生了显著变化,由线性混合模型(LMM)确定。热图行注释显示了由LMMs(基线 vs. W2和基线 vs. W12)和多元回归模型(MDD vs HC)得到的有色系数。系数有不同的颜色,蓝色(下调或富集于HC)和红色(上调或富集于MDD)。显著差异由p<0.05,* p<0.01,*** q<0.1表示。
d)6种血浆代谢物(来自c)的纵向变化,它们在R和NR组之间表现出不同的趋势。P值由应用于R和NR组数据的LMMs得到。
2、ESC治疗对肠道菌群有抑制作用
接下来,我们关注了ESC对肠道微生物的影响。如图3A所示,ESC治疗显著降低了物种水平的丰富度,但没有降低基因水平的丰富度。在亚组分析中,我们观察到微生物丰富度的降低在NR组从基线到第12周特别明显。然而,需要注意的是,在基因水平上,从基线到第2周观察到了丰富度的暂时增加。在R组,物种或基因水平的丰富度没有显著变化。此外,R组的丰富度在基线和第12周都显著高于NR组,但始终低于HC组。与丰富度分析类似,我们在Shannon指数(图S2A)中也观察到了类似的趋势。这些结果提醒我们,ESC对肠道微生物的影响在R和NR组之间,以及在短期(基线到第2周)和长期(基线到第12周)治疗期间有所不同。
PERMANOVA结果显示,肠道微生物组成在第12周发生了显著变化(约束PERMANOVA,R2=0.0046,p=0.034),但在第2周没有变化(图3B)。我们的观察表明,即使在第12周,MDD患者和HC组的肠道微生物组成之间仍然存在明显的差异(Bray–Curtis距离,R2=0.027,p=0.00033)。这表明ESC治疗并没有有效地将MDD患者的肠道微生物组转变为更健康或更接近HC组的状态。然而,HAMD-17评分与PCoA1呈负相关(LMM,系数β=-3.39,p=0.05),这表明抑郁症状的缓解与肠道微生物的变化有显著的关联。为了进一步探索MDD患者的肠道微生物变化是否朝着更健康的状态发展,我们使用HC样本作为参照,为每个MDD样本估计了一个菌群失调评分(DS),并计算了MDD和HC队列之间的Bray–Curtis距离的中位数。我们的发现显示,DS与物种丰富度呈显著的负相关(图S2B)。总体而言,DS从基线到第12周没有显著改变(p=0.276)。然而,DS在NR组高于R组,在基线和第12周都有显著差异,其中第12周的差异最为显著(p=0.031;图3C)。根据这些发现,ESC并没有改善整体的肠道微生物状况,但NR组的肠道微生物比R组更为紊乱。
在个体分类水平上,我们的分析显示,ESC治疗后,特定物种发生了显著的变化。具体来说,从基线到第2周,我们观察到五个物种的促进和七个物种的抑制。同样地,从基线到第12周,ESC治疗导致了六个物种的促进和九个物种的抑制(图3D)。
在微生物功能方面,使用MaAsLin2分析了代谢途径的变化,以受试者为随机效应。ESC治疗后,所有样本中有145条途径发生了显著改变,其中143条途径被下调(q<0.25)。进一步的亚组分析显示,NR组中有17条途径被下调,而R组中没有途径发生改变(使用显著性阈值q<0.25,图3E)。考虑到SSRIs 的已知抗菌特性,我们还确定了ESC对抗菌耐药性(AMR)基因的影响。我们的发现显示,ESC治疗后,AMR基因呈现出整体上调的趋势。具体来说,在分析的15个ESC改变的基因中,有11个基因表现出上调,使用显著性截止值p<0.05 (表S5)。
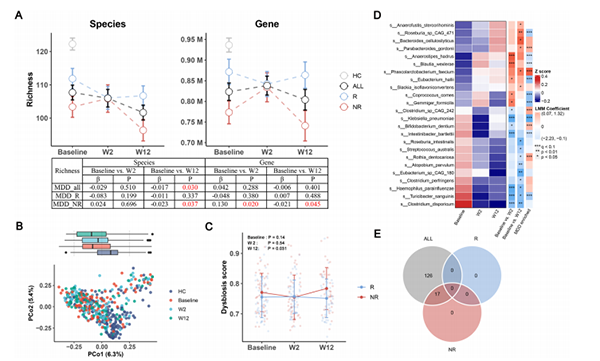
图3 ESC治疗对肠道菌群的影响。
a) 在ALL、R和NR组中,从基线到第12周,物种和基因水平上的微生物丰度的变化,以HC为参照。ALL包括了R和NR组的样本。表格展示了使用LMM进行纵向比较的详细结果,包括访问周系数和P值。
b )基于不同组的宏基因组谱的主坐标分析(PCoA)图。
c ) R和NR组中,从基线到第12周,菌群失调评分(DS)的变化。NR组在第12周显著高于R组(P=0.031)。
d ) 热图显示了24个物种的平均丰度,这些物种在ESC干预后的第2周或第12周发生了显著变化,由线性混合模型确定。热图行注释显示了由LMMs(基线 vs. W2和基线 vs. W12)和多元回归模型(MDD vs HC)得到的有色系数。系数有不同的颜色,蓝色(在HC中被抑制或富集)和红色(在MDD中被促进或富集)。显著差异用 p<0.05, * p<0.01, *** q<0.1表示。
e) ALL(q<0.25)、R(q<0.25)和NR(q<0.25)组在ESC干预后显著下调的metaCyc途径的韦恩图。
4、血浆代谢组和肠道菌群的整合网络
前述发现表明,ESC治疗影响了肠道菌群和血浆代谢组。这些结果与以前的发现一致,表明肠道菌群可以在调节个体血浆代谢物谱方面发挥作用,其影响程度根据特定的疾病或状况而异。因此,我们继续探索两组组学数据之间的关联。我们首先使用HC和MDD队列的样本匹配的组学数据进行了一项Procrustes分析。结果显示,MDD患者(Monte Carlo P值=0.013)和HC(Monte Carlo P值=0.092;图4A)的血浆代谢组和肠道菌群之间存在弱的对应关系。正如预期的那样,我们观察到在MDD(Monte Carlo P值=0.0445)和HC(Monte Carlo P值=5e-04)队列中,粪便代谢组和肠道菌群之间存在更强的相关性(图S3A)。鉴于血浆代谢组和肠道菌群之间缺乏显著的整体关联,我们推测这种关联更可能发生在代谢物和肠道菌群之间的个体水平,而不是整体的组学关联。
另外,我们的分析显示,MDD和HC组之间,肠道菌群解释的血浆代谢物的方差存在显著差异,MDD人群中解释的方差更高(图4B)。这些结果表明,与健康对照相比肠道微生物对MDD个体的血浆代谢物有更强的影响。总体而言,在血浆代谢物和肠道菌群之间的关联中,检测到了MDD特异性的模式。
接下来,为了进一步建立血浆代谢物和肠道菌群之间的关联,我们使用所有MDD样本构建了glmmLasso模型。对于每一种代谢物,拟合glmmLasso模型来选择可能相关的细菌物种,然后将这些选定的特征进一步拟合到LMM模型中,以评估其显著性水平(见方法)。在FDR校正后(q<0.1,表S9),发现了109种代谢物和139种物种之间的255个显著关联。如图4C和表S9所示,19种被ESC治疗改变的代谢物与细菌物种相关,而上调的代谢物的数量远高于下调的代谢物。例如,L-色氨酸与Parabacteroides goldsteinii和Streptococcus thermophilus相关。后者属于色氨酸代谢细菌,含有可以通过色氨酸代谢维持细菌生长和存活的色氨酸酶,这些细菌产生的代谢物可以调节微生物多样性并给宿主带来益处。在我们的分析中,我们发现I3PA与Bacteroides plebeius和Streptococcus cristatus相关。此外,我们观察到I3PA与物种丰富度显著正相关(图4D)。综上所述,以上发现表明,在ESC治疗后,血浆代谢物和肠道微生物之间存在潜在的相关性,其中氨基酸代谢和色氨酸衍生物可能发挥重要作用。
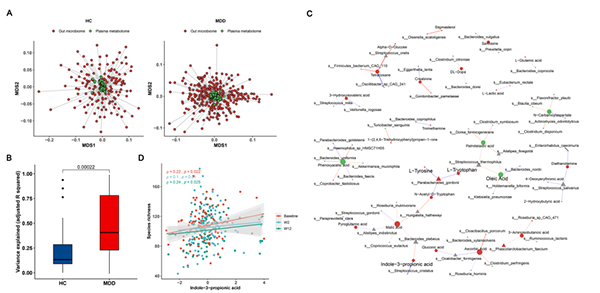
图4 血浆代谢组和肠道菌群之间的关联。
a )Procrustes分析的多维缩放(MDS)图,显示了MDD和HC队列中血浆代谢组和肠道菌群的整体关联,个体样本用线连接。对血浆代谢组(绿色圆圈)使用欧氏距离,对肠道菌群数据(红色圆圈)使用Bray–Curtis距离,并标注了Procrustes m2统计结果。
b )与HC相比,MDD患者中肠道菌群解释的血浆代谢物的方差显著更高。使用lasso模型得到的r.2
统计量来量化解释的方差程度。
c)由glmmLasso模型生成的血浆代谢物和微生物物种之间的关联网络。分析只展示了那些对ESC治疗有显著反应的代谢物。节点大小表示度数,不同的形状表示代谢物(圆圈)和微生物(三角形),节点的不同颜色表示ESC干预后的上调(红色)和下调(绿色),负相关用蓝色表示,正相关用红色表示。
d) 散点图显示了三个访问周的物种丰富度和吲哚-3-丙酸(I3PA)之间的斯皮尔曼相关性。显著的正相关只在基线和第12周观察到。
5、不同缓解组的基线微生物组特征
我们研究的主要目的之一是探讨肠道菌群对抗抑郁药物效果的预测潜力。为了达到这个目的,我们试图比较基线时R和NR组之间的微生物组特征。尽管PERMANOVA分析显示,两个亚组在基线时的微生物组成没有显著差异(p=0.51;图5A),但R组的微生物丰富度在物种(p=0.017)和基因(p=0.019)水平上都显著高于NR组(图5B)。此外,物种丰富度与基线时的HAMD-17呈负相关(图S4A)。Shannon指数也呈现出类似的趋势(p=0.082,p=0.002),尽管在物种水平上没有显著性(图S4B)。事实上,肠道菌群的多样性被报道能够在促进微生物恢复力和增强其稳定性方面发挥积极作用 。为了进一步估计微生物群落的恢复力,我们使用sparCC为R和NR组构建了微生物相互作用网络,并发现R组的群落结构和细菌网络比NR组更复杂和更有组织(图S4C)。然后,我们通过随机移除节点来模拟对网络的“生态攻击” ,比较了两个网络的恢复力。自然连通性被用来评估剩余网络的鲁棒性。尽管在“攻击”下,R网络的自然连通性始终高于NR网络(图S4D)。这些结果支持了R组中更有恢复力的细菌群落。此外,我们观察到,几个产孢类群在R组中比NR组更丰富,包括Clostridiaceae、Eubacterium rectale和C. comes(P<0.05;图5C)。产孢细菌可以抵抗外界环境刺激,促进肠道菌群重建,并恢复肠道稳态。值得注意的是,E. rectale,一种核心的肠道共生物种,在R组和NR组的三个访问周中,始终表现出更高的丰度,并且其丰度与基线和第12周的物种丰富度呈正相关(图S4E&F)。作为一种严格的厌氧菌,E. rectale可以通过从不可消化的纤维中产生丁酸盐和其他SCFAs来促进宿主肠道健康,并且最近被确定为心理生物,以维持和改善心理健康 。与产孢细菌的情况类似,大多数孢子形成基因(在至少20%的基线样本中出现的22个孢子形成基因中的11个)在R组中比NR组更高(q<0.1,图5D)。此外,22个孢子形成基因中的18个与物种丰富度呈显著正相关(表S10)。因此,我们推测,微生物孢子形成可能有助于肠道菌群对ESC诱导的扰动的抵抗。
为了更好地理解基线时R和NR组之间的微生物差异,我们使用Wilcox检验比较了两个亚组之间的非冗余基因的丰度,并得到了32,231个差异基因(p<0.01)。差异基因被聚类成共丰度组(CAGs),并使用canopy聚类算法进行了分类学注释。如图5E所示,大多数差异基因被注释为Clostridales(78.40%)。与NR组相比,CAG注释的物种,包括产丁酸菌Ruminococcus lactaris、Faecaecalibacterium prausnitzii、E. rectale和Romboutsia在R组中富集。丁酸盐对肠道平衡有积极的影响,通过抑制炎症和癌变,增强结肠防御屏障的各个组成部分,减少氧化应激。此外,CAG注释的有益细菌,如Bifdobacterium animalis和Lactobacillus sanfranciscens,也在R组中富集。我们的结果支持了这样一个观点,即R组的整体肠道菌群状况可能比NR组的基线时更好。

图5 基线时R和NR组之间的微生物组比较。
a)使用Bray–Curtis差异度的PCoA图,显示R和NR组之间的微生物组成没有显著差异(PERMANOVA P=0.5)。
b)R和NR组之间的微生物物种(左)和基因(右)的丰富度差异,以HC为参照。
c) 缓解组中肠道微生物物种的回归系数的火山图。使用多元回归模型探索肠道菌群和临床缓解之间的关联。p值<0.05被认为是统计显著的。在R组中,丰度较高的物种用蓝色点表示,而丰度较低的物种用红色点表示。
d)显示22个孢子形成基因的平均丰富度的热图,其中11个在R组中富集,由多元回归模型确定。热图行注释显示了由MLR模型(R vs NR)得到的有色系数。系数有不同的颜色,蓝色(在NR中富集)和红色(在R中富集)。显著差异用* p<0.05表示。
e ) R和NR组之间在基线时差异丰富的基因的共丰度组(CAGs)的热图。基因丰富度由颜色渐变表示(红色表示最高丰富度,白色表示零,蓝色表示最低丰富度)
6、产孢基因作为ESC反应中MDD缓解的预测因子
为了探索基线时肠道菌群对MDD患者治疗缓解的预测潜力,我们使用从基线样本中得到的三种不同的特征集(分类单元、多样性和孢子形成基因)构建了预测模型。为了确保模型性能的无偏评估,我们在本研究中采用了嵌套交叉验证(图S5A,见方法)。嵌套交叉验证是一种将模型选择和超参数优化过程结合在评估过程中的技术。使用接收者操作特征(ROC)曲线下的面积(AUC)来评估模型性能。在我们的建模结果中,微生物分类单元表现出低性能(平均AUC=0.61;图6A)。然而,孢子形成基因模型(平均AUC=0.736)与十个外循环模型(AUC=0.710,通过合并所有外循环测试结果绘制的ROC)的组合,达到了最高的平均预测性能(图6B)。考虑到ESC对孢子形成基因的潜在影响,我们还根据从基线到第2周的孢子形成基因的变化,预测了MDD患者的临床缓解。结果表明,平均AUC和联合AUC分别为0.734和0.701,表明模型具有中等到良好的预测性能(图S5B)。
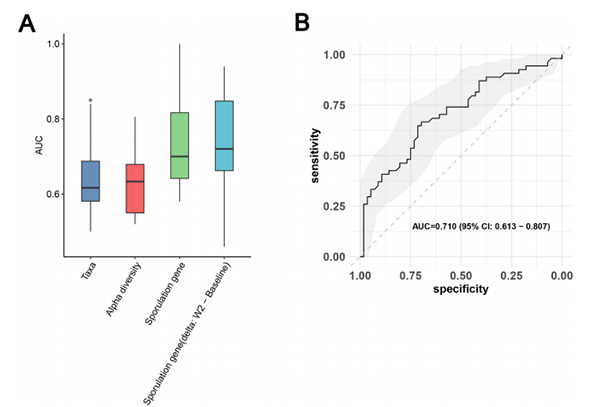
图6 基线时肠道微生物组特征对MDD患者临床缓解的预测价值。
a) 每个特征集的10个外循环测试结果的AUC箱线图,使用随机森林训练数据,平均AUC从0.56到0.73变化(分类单元:AUC=0.65;Alpha多样性:AUC=0.63;芽孢形成基因:AUC=0.736;W2和基线之间的芽孢形成基因差值:AUC=0.734)。
b) 基于芽孢形成基因的RF模型的10个外循环测试结果的连接ROC曲线
结论
总之,通过对MDD患者进行纵向研究,我们对多组学数据进行了全面分析,以探讨肠道菌群在ESC治疗后缓解中的作用。我们对血液代谢组的分析显示,ESC治疗改善了异常的血液代谢,通过上调MDD缺乏的氨基酸和下调MDD富集的脂肪酸。此外,ESC对肠道菌群产生了弱抑制效应,但是缓解者由于物种丰富度和芽孢形成机制的高,表现出比非缓解者更强的微生物群落稳定性。另外,我们的发现表明,肠道菌群在塑造血液代谢组的变异性方面发挥作用,并与几种代谢物(例如L-色氨酸和I3PA)相关,这些代谢物在ESC治疗后上调。我们的数据支持了这样一个观点,改善的肠道菌群配置可以促进抗抑郁治疗的有效性,特别是在ESC的情况下。总的来说,这项研究为抗抑郁药物、血液代谢组改变和MDD患者肠道菌群之间的相互关联关系提供了新的见解。通过揭示这些机制,我们的发现有助于增进我们对ESC在缓解抑郁方面发挥其治疗效果的方式的理解。此外,这些知识可以通过针对肠道菌群和调节相关的血液代谢组,帮助开发改善的MDD治疗策略。
本文译自:
Yaping Wang , Jingjing Zhou , Junbin Ye et al. Multi-omics reveal microbial determinants impacting the treatment outcome of antidepressants in major depressive disorder. Microbiome. 2023 Aug 28; 11(1): 195.
